Cu2+(MeOH)n=9,10团簇的势能面探索:溶剂相与气相
IF 2
3区 化学
Q4 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
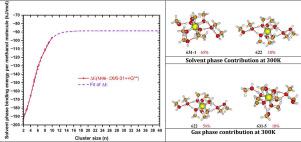
在本工作中,我们研究了两种不同介质:气相和溶剂相中甲醇簇中Cu2+离子结构的势能面(PESs)。利用M06-2X/6-31++G(d,p)理论水平,利用积分方程形式极化连续体模型(IEF-PCM)研究了介质极化对Cu2+(MeOH)n=9,10团簇结构和能能参数的影响。因此,在溶剂相中,簇结构在全局最小同分异构体中是六配位的。对温度依赖性的研究表明,六、五配位结构在溶剂相中与具有两个溶剂化壳层的六配位结构竞争。离子键的Wiberg键指数(WBI)分析证实了这一结构研究。与气体介质相比,在IEF-PCM溶剂中,四、五坐标构象的Wiberg键指数较弱,而六坐标构象在两种介质中的Wiberg键指数几乎相同。这证明了与溶剂相相比,气相中的结合键更强。饱和时的电子结合能在单一拟合函数下分别为-88.6和-146.6 kJmol−1,这也支持了这一点。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Exploration of the potential energy surfaces of the Cu2+(MeOH)n=9,10 clusters: Solvent phase vs gas phase
In the present work, we investigated the potential energy surfaces (PESs) of the structures of the ion in methanol clusters in two different media: gas and solvent phases. The effects of medium polarization by the integral equation formalism polarized continuum model (IEF-PCM) on structural and energetic parameters were examined on the conformers of the clusters using the M06-2X/6-31++G(d,p) level of theory. Thus, in the solvent phase, the cluster structures are hexa-coordinated in the global minimum isomer. The study of the temperature dependency shows that the hexa-, and penta-coordinated structures compete with a large predominance of the hexa-coordinate structures with two solvation shells in the solvent phase. The Wiberg bond indices (WBI) analysis of the ionic bond confirms the structural study. In the IEF-PCM solvent compared to the gas medium, Wiberg bond indices of the tetra-, and penta-coordinate conformers are weaker, and the hexa-coordinate conformers in both media are nearly identical. This proves that compared to the solvent phase, the dative bond is stronger in the gas phase. This is supported by the significant difference in the electronic binding energies at saturation found with a single fitting function which are -88.6 and -146.6 in the solvent and gas phases, respectively.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Chemical Physics
化学-物理:原子、分子和化学物理
CiteScore
4.60
自引率
4.30%
发文量
278
审稿时长
39 days
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


