模拟镁的位错行为的机器学习潜力
IF 15.8
1区 材料科学
Q1 METALLURGY & METALLURGICAL ENGINEERING
引用次数: 0
摘要
通过分子动力学(MD)模拟准确预测镁(Mg)的位错行为对于研究变形的基本机制和设计高塑性镁合金至关重要。然而,MD 模拟中现有的镁原子势对于许多位错相关现象(如堆积断层能(SFE)和位错核心结构)的定量不够。在这里,通过结合 468 个密度泛函理论(DFT)计算数据点和一种机器学习方法,我们创建了一种广泛适用的深度学习势(DLP)来研究镁的位错行为。我们证明,我们的 DLP 重现了 SFE、晶格常数、弹性常数和表面能,与实验或 DFT 数据基本一致。此外,DLP 预测的基性〈a〉、棱柱形〈a〉、金字塔形〈c + a〉位错都与 DFT 关于解离距离和核心结构的结果十分吻合。重要的是,DLP在区分金字塔形〈I〉和〈II〉〈c + a〉螺位错核心结构方面表现出色。我们的研究结果表明,DLP 适用于研究镁的位错行为,这使其对未来一般变形问题的现实原子研究具有重要价值。本文章由计算机程序翻译,如有差异,请以英文原文为准。
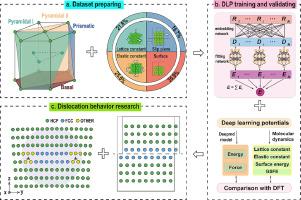
A machine learning potential for simulation the dislocation behavior of magnesium
Accurate predictions of the dislocation behavior of magnesium (Mg) by molecular dynamics (MD) simulations are essential for studying the fundamental mechanisms of deformation and designing high plasticity Mg alloys. However, existing atomic potentials in MD simulation for Mg are not sufficiently quantitative for many dislocations-associated phenomena, such as stacking fault energy (SFE) and dislocation core structures. Here, by combining 468 density functional theory (DFT) calculated data points and a machine learning method, we create a broadly applicable deep learning potential (DLP) to study the dislocation behavior of Mg. We demonstrate that our DLP reproduces the SFE, lattice constants, elastic constants, and surface energies in reasonable agreement with experimental or DFT data. Furthermore, the DLP predicted basal 〈a〉, prismatic 〈a〉, pyramidal 〈c + a〉 dislocations all agree well with DFT results on dissociation distance and core structures. Importantly, the DLP has a superior performance on distinguishing the pyramidal I and II 〈c + a〉 screw dislocation core structures. Our results show that the DLP is suitable for investigating the dislocation behavior of Mg, making it valuable for future realistic atomistic studies of general deformation problems.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Magnesium and Alloys
Engineering-Mechanics of Materials
CiteScore
20.20
自引率
14.80%
发文量
52
审稿时长
59 days
期刊介绍:
The Journal of Magnesium and Alloys serves as a global platform for both theoretical and experimental studies in magnesium science and engineering. It welcomes submissions investigating various scientific and engineering factors impacting the metallurgy, processing, microstructure, properties, and applications of magnesium and alloys. The journal covers all aspects of magnesium and alloy research, including raw materials, alloy casting, extrusion and deformation, corrosion and surface treatment, joining and machining, simulation and modeling, microstructure evolution and mechanical properties, new alloy development, magnesium-based composites, bio-materials and energy materials, applications, and recycling.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


