Christina E. Costa, Marina M. Watowich, Elisabeth A. Goldman, Kirstin N. Sterner, Josue E. Negron-Del Valle, Daniel Phillips, Cayo Biobank Research Unit, Michael L. Platt, Michael J. Montague, Lauren J. N. Brent, James P. Higham, Noah Snyder-Mackler, Amanda J. Lea
{"title":"猕猴免疫细胞 DNA 甲基化的遗传结构","authors":"Christina E. Costa, Marina M. Watowich, Elisabeth A. Goldman, Kirstin N. Sterner, Josue E. Negron-Del Valle, Daniel Phillips, Cayo Biobank Research Unit, Michael L. Platt, Michael J. Montague, Lauren J. N. Brent, James P. Higham, Noah Snyder-Mackler, Amanda J. Lea","doi":"10.1111/mec.17576","DOIUrl":null,"url":null,"abstract":"<p>Genetic variation that impacts gene regulation, rather than protein function, can have strong effects on trait variation both within and between species. Epigenetic mechanisms, such as DNA methylation, are often an important intermediate link between genotype and phenotype, yet genetic effects on DNA methylation remain understudied in natural populations. To address this gap, we used reduced representation bisulfite sequencing to measure DNA methylation levels at 555,856 CpGs in peripheral whole blood of 573 samples collected from free-ranging rhesus macaques (<i>Macaca mulatta</i>) living on the island of Cayo Santiago, Puerto Rico. We used allele-specific methods to map <i>cis</i>-methylation quantitative trait loci (meQTL) and tested for effects of 243,389 single nucleotide polymorphisms (SNPs) on local DNA methylation levels. Of 776,092 tested SNP–CpG pairs, we identified 516,213 meQTL, with 69.12% of CpGs having at least one meQTL (FDR < 5%). On average, meQTL explained 21.2% of nearby methylation variance, significantly more than age or sex. meQTL were enriched in genomic compartments where methylation is likely to impact gene expression, for example, promoters, enhancers and binding sites for methylation-sensitive transcription factors. In support, using mRNA-seq data from 172 samples, we confirmed 332 meQTL as whole blood <i>cis</i>-expression QTL (eQTL) in the population, and found meQTL–eQTL genes were enriched for immune response functions, like antigen presentation and inflammation. Overall, our study takes an important step towards understanding the genetic architecture of DNA methylation in natural populations, and more generally points to the biological mechanisms driving phenotypic variation in our close relatives.</p>","PeriodicalId":210,"journal":{"name":"Molecular Ecology","volume":"34 15","pages":""},"PeriodicalIF":3.9000,"publicationDate":"2024-11-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12269644/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genetic Architecture of Immune Cell DNA Methylation in the Rhesus Macaque\",\"authors\":\"Christina E. Costa, Marina M. Watowich, Elisabeth A. Goldman, Kirstin N. Sterner, Josue E. Negron-Del Valle, Daniel Phillips, Cayo Biobank Research Unit, Michael L. Platt, Michael J. Montague, Lauren J. N. Brent, James P. Higham, Noah Snyder-Mackler, Amanda J. Lea\",\"doi\":\"10.1111/mec.17576\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Genetic variation that impacts gene regulation, rather than protein function, can have strong effects on trait variation both within and between species. Epigenetic mechanisms, such as DNA methylation, are often an important intermediate link between genotype and phenotype, yet genetic effects on DNA methylation remain understudied in natural populations. To address this gap, we used reduced representation bisulfite sequencing to measure DNA methylation levels at 555,856 CpGs in peripheral whole blood of 573 samples collected from free-ranging rhesus macaques (<i>Macaca mulatta</i>) living on the island of Cayo Santiago, Puerto Rico. We used allele-specific methods to map <i>cis</i>-methylation quantitative trait loci (meQTL) and tested for effects of 243,389 single nucleotide polymorphisms (SNPs) on local DNA methylation levels. Of 776,092 tested SNP–CpG pairs, we identified 516,213 meQTL, with 69.12% of CpGs having at least one meQTL (FDR < 5%). On average, meQTL explained 21.2% of nearby methylation variance, significantly more than age or sex. meQTL were enriched in genomic compartments where methylation is likely to impact gene expression, for example, promoters, enhancers and binding sites for methylation-sensitive transcription factors. In support, using mRNA-seq data from 172 samples, we confirmed 332 meQTL as whole blood <i>cis</i>-expression QTL (eQTL) in the population, and found meQTL–eQTL genes were enriched for immune response functions, like antigen presentation and inflammation. Overall, our study takes an important step towards understanding the genetic architecture of DNA methylation in natural populations, and more generally points to the biological mechanisms driving phenotypic variation in our close relatives.</p>\",\"PeriodicalId\":210,\"journal\":{\"name\":\"Molecular Ecology\",\"volume\":\"34 15\",\"pages\":\"\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-11-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12269644/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Ecology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/mec.17576\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Ecology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/mec.17576","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
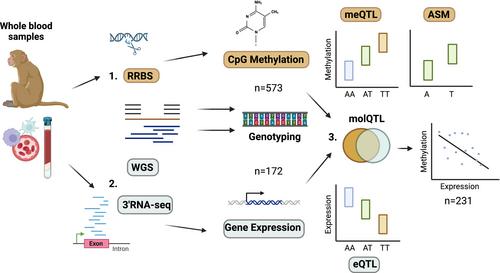
影响基因调控而非蛋白质功能的遗传变异会对物种内和物种间的性状变异产生强烈影响。表观遗传机制(如 DNA 甲基化)往往是基因型和表型之间的重要中间环节,但在自然种群中,遗传对 DNA 甲基化的影响仍未得到充分研究。为了填补这一空白,我们使用还原表征亚硫酸氢盐测序法测量了从生活在波多黎各卡约圣地亚哥岛上的散养猕猴采集的 573 份外周全血样本中 555,856 个 CpGs 的 DNA 甲基化水平。我们使用等位基因特异性方法绘制了顺式甲基化定量性状位点(meQTL)图,并检测了 243,389 个单核苷酸多态性(SNPs)对当地 DNA 甲基化水平的影响。在 776,092 个受测 SNP-CpG 对中,我们发现了 516,213 个 meQTL,其中 69.12% 的 CpGs 至少有一个 meQTL(FDR
Genetic Architecture of Immune Cell DNA Methylation in the Rhesus Macaque
Genetic variation that impacts gene regulation, rather than protein function, can have strong effects on trait variation both within and between species. Epigenetic mechanisms, such as DNA methylation, are often an important intermediate link between genotype and phenotype, yet genetic effects on DNA methylation remain understudied in natural populations. To address this gap, we used reduced representation bisulfite sequencing to measure DNA methylation levels at 555,856 CpGs in peripheral whole blood of 573 samples collected from free-ranging rhesus macaques (Macaca mulatta) living on the island of Cayo Santiago, Puerto Rico. We used allele-specific methods to map cis-methylation quantitative trait loci (meQTL) and tested for effects of 243,389 single nucleotide polymorphisms (SNPs) on local DNA methylation levels. Of 776,092 tested SNP–CpG pairs, we identified 516,213 meQTL, with 69.12% of CpGs having at least one meQTL (FDR < 5%). On average, meQTL explained 21.2% of nearby methylation variance, significantly more than age or sex. meQTL were enriched in genomic compartments where methylation is likely to impact gene expression, for example, promoters, enhancers and binding sites for methylation-sensitive transcription factors. In support, using mRNA-seq data from 172 samples, we confirmed 332 meQTL as whole blood cis-expression QTL (eQTL) in the population, and found meQTL–eQTL genes were enriched for immune response functions, like antigen presentation and inflammation. Overall, our study takes an important step towards understanding the genetic architecture of DNA methylation in natural populations, and more generally points to the biological mechanisms driving phenotypic variation in our close relatives.
期刊介绍:
Molecular Ecology publishes papers that utilize molecular genetic techniques to address consequential questions in ecology, evolution, behaviour and conservation. Studies may employ neutral markers for inference about ecological and evolutionary processes or examine ecologically important genes and their products directly. We discourage papers that are primarily descriptive and are relevant only to the taxon being studied. Papers reporting on molecular marker development, molecular diagnostics, barcoding, or DNA taxonomy, or technical methods should be re-directed to our sister journal, Molecular Ecology Resources. Likewise, papers with a strongly applied focus should be submitted to Evolutionary Applications. Research areas of interest to Molecular Ecology include:
* population structure and phylogeography
* reproductive strategies
* relatedness and kin selection
* sex allocation
* population genetic theory
* analytical methods development
* conservation genetics
* speciation genetics
* microbial biodiversity
* evolutionary dynamics of QTLs
* ecological interactions
* molecular adaptation and environmental genomics
* impact of genetically modified organisms

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


