非苯类芳香烃的表面化学键:烯与芘的对比
IF 2.1
4区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
缺陷对碳基(光)电子材料的性能起着至关重要的作用,因为材料与金属电极的相互作用在很大程度上取决于π电子系统的拓扑结构。然而,由于缺陷的密度通常很低,因此很难对缺陷进行直接研究。为了解决这个问题,我们使用了一个分子模型系统,比较了多环芳烃芘及其异构体醋楹与铜(111)表面的相互作用。醋楹是一种具有非苯类非交替拓扑结构的模型缺陷,而芘则代表了一种理想的苯类交替结构。我们发现,醋楹与金属表面形成的结合比芘更牢固。这表现在分子-表面键能更高、吸附引起的电子结构变化显著(通过光电子和 X 射线吸收光谱进行研究)以及吸附高度可能更低(根据非接触式原子力显微镜)。醋螨烯的键更强与它的最高占位轨道和最低未占位轨道之间的间隙(HOMO-LUMO 间隙)更小有关,这使得 LUMO 更接近金属的费米能,从而与金属的电子状态产生更强的杂化。密度泛函理论计算支持我们的发现,表明非苯类、非替代结构元素可以增强石墨烯基材料与金属电极之间的结合。此外,这些结果突显了非类苯分子有机半导体作为类苯分子有机半导体替代品的潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
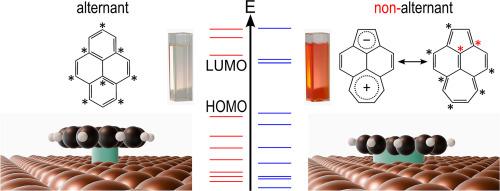
The surface chemical bond of a nonbenzenoid aromatic hydrocarbon: Acepleiadylene versus pyrene
Defects play a critical role in the performance of carbon-based (opto-)electronic materials, because the materials’ interaction with metal electrodes can strongly depend on the topology of the π-electron system. However, the direct investigation of defects is difficult due to their typically low density. To address this issue, we use a molecular model system comparing the polycyclic aromatic hydrocarbon pyrene with its isomer acepleiadylene regarding their interaction with a Cu(111) surface. Acepleiadylene serves as a model defect with a nonbenzenoid nonalternant topology, while pyrene represents an ideal benzenoid alternant structure. We find that acepleiadylene forms a stronger bond to the metal surface than pyrene. This is evidenced by a higher molecule-surface bond energy, significant adsorption-induced changes in electronic structure (studied via photoelectron and X-ray absorption spectroscopies), and a potentially lower adsorption height (according to non-contact atomic force microscopy). The stronger bond of acepleiadylene is linked to its smaller gap between the highest occupied and the lowest unoccupied orbitals (HOMO-LUMO gap), bringing the LUMO closer to the metal's Fermi energy and resulting in stronger hybridization with the metal's electronic states. Density functional theory calculations support our findings, suggesting that nonbenzenoid, nonalternant structural elements can enhance the bonding between graphene-based materials and metal electrodes. Additionally, these results highlight the potential of nonbenzenoid molecular organic semiconductors as alternatives to their benzenoid counterparts.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Surface Science
化学-物理:凝聚态物理
CiteScore
3.30
自引率
5.30%
发文量
137
审稿时长
25 days
期刊介绍:
Surface Science is devoted to elucidating the fundamental aspects of chemistry and physics occurring at a wide range of surfaces and interfaces and to disseminating this knowledge fast. The journal welcomes a broad spectrum of topics, including but not limited to:
• model systems (e.g. in Ultra High Vacuum) under well-controlled reactive conditions
• nanoscale science and engineering, including manipulation of matter at the atomic/molecular scale and assembly phenomena
• reactivity of surfaces as related to various applied areas including heterogeneous catalysis, chemistry at electrified interfaces, and semiconductors functionalization
• phenomena at interfaces relevant to energy storage and conversion, and fuels production and utilization
• surface reactivity for environmental protection and pollution remediation
• interactions at surfaces of soft matter, including polymers and biomaterials.
Both experimental and theoretical work, including modeling, is within the scope of the journal. Work published in Surface Science reaches a wide readership, from chemistry and physics to biology and materials science and engineering, providing an excellent forum for cross-fertilization of ideas and broad dissemination of scientific discoveries.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


