{"title":"电子传递紊乱:新形式的类固醇生成缺陷和线粒体病。","authors":"Walter L Miller, Amit V Pandey, Christa E Flück","doi":"10.1210/clinem/dgae815","DOIUrl":null,"url":null,"abstract":"<p><p>Most disorders of steroidogenesis, such as forms of congenital adrenal hyperplasia (CAH) are caused by mutations in genes encoding the steroidogenic enzymes and are often recognized clinically by cortisol deficiency, hyper- or hypo-androgenism, and/or altered mineralocorticoid function. Most steroidogenic enzymes are forms of cytochrome P450. Most P450s, including several steroidogenic enzymes, are microsomal, requiring electron donation by P450 oxidoreductase (POR); however, several steroidogenic enzymes are mitochondrial P450s, requiring electron donation via ferredoxin reductase (FDXR) and ferredoxin (FDX). POR deficiency is a rare but well-described form of CAH characterized by impaired activity of 21-hydroxylase (P450c21, CYP21A2) and 17-hydroxylase/17,20-lyase (P450c17, CYP17A1); more severely affected individuals also have the Antley-Bixler skeletal malformation syndrome and disordered genital development in both sexes, and hence is easily recognized. The 17,20-lyase activity of P450c17 requires both POR and cytochrome b5 (b5), which promote electron transfer. Mutations of POR, b5, or P450c17 can cause selective 17,20-lyase deficiency. In addition to providing electrons to mitochondrial P450s, FDX, and FDXR are required for the synthesis of iron-sulfur clusters, which are used by many enzymes. Recent work has identified FDXR mutations in patients with visual impairment, optic atrophy, neuropathic hearing loss, and developmental delay, resembling the global neurologic disorders seen with mitochondrial diseases. Many of these patients have had life-threatening events or deadly infections, often without an apparent triggering event. Adrenal insufficiency has been predicted in such individuals but has only been documented recently. Neurologists, neonatologists, and geneticists should seek endocrine assistance in evaluating and treating patients with mutations in FDXR.</p>","PeriodicalId":50238,"journal":{"name":"Journal of Clinical Endocrinology & Metabolism","volume":" ","pages":"e574-e582"},"PeriodicalIF":5.1000,"publicationDate":"2025-02-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11834722/pdf/","citationCount":"0","resultStr":"{\"title\":\"Disordered Electron Transfer: New Forms of Defective Steroidogenesis and Mitochondriopathy.\",\"authors\":\"Walter L Miller, Amit V Pandey, Christa E Flück\",\"doi\":\"10.1210/clinem/dgae815\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Most disorders of steroidogenesis, such as forms of congenital adrenal hyperplasia (CAH) are caused by mutations in genes encoding the steroidogenic enzymes and are often recognized clinically by cortisol deficiency, hyper- or hypo-androgenism, and/or altered mineralocorticoid function. Most steroidogenic enzymes are forms of cytochrome P450. Most P450s, including several steroidogenic enzymes, are microsomal, requiring electron donation by P450 oxidoreductase (POR); however, several steroidogenic enzymes are mitochondrial P450s, requiring electron donation via ferredoxin reductase (FDXR) and ferredoxin (FDX). POR deficiency is a rare but well-described form of CAH characterized by impaired activity of 21-hydroxylase (P450c21, CYP21A2) and 17-hydroxylase/17,20-lyase (P450c17, CYP17A1); more severely affected individuals also have the Antley-Bixler skeletal malformation syndrome and disordered genital development in both sexes, and hence is easily recognized. The 17,20-lyase activity of P450c17 requires both POR and cytochrome b5 (b5), which promote electron transfer. Mutations of POR, b5, or P450c17 can cause selective 17,20-lyase deficiency. In addition to providing electrons to mitochondrial P450s, FDX, and FDXR are required for the synthesis of iron-sulfur clusters, which are used by many enzymes. Recent work has identified FDXR mutations in patients with visual impairment, optic atrophy, neuropathic hearing loss, and developmental delay, resembling the global neurologic disorders seen with mitochondrial diseases. Many of these patients have had life-threatening events or deadly infections, often without an apparent triggering event. Adrenal insufficiency has been predicted in such individuals but has only been documented recently. Neurologists, neonatologists, and geneticists should seek endocrine assistance in evaluating and treating patients with mutations in FDXR.</p>\",\"PeriodicalId\":50238,\"journal\":{\"name\":\"Journal of Clinical Endocrinology & Metabolism\",\"volume\":\" \",\"pages\":\"e574-e582\"},\"PeriodicalIF\":5.1000,\"publicationDate\":\"2025-02-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11834722/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Endocrinology & Metabolism\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1210/clinem/dgae815\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Endocrinology & Metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1210/clinem/dgae815","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Disordered Electron Transfer: New Forms of Defective Steroidogenesis and Mitochondriopathy.
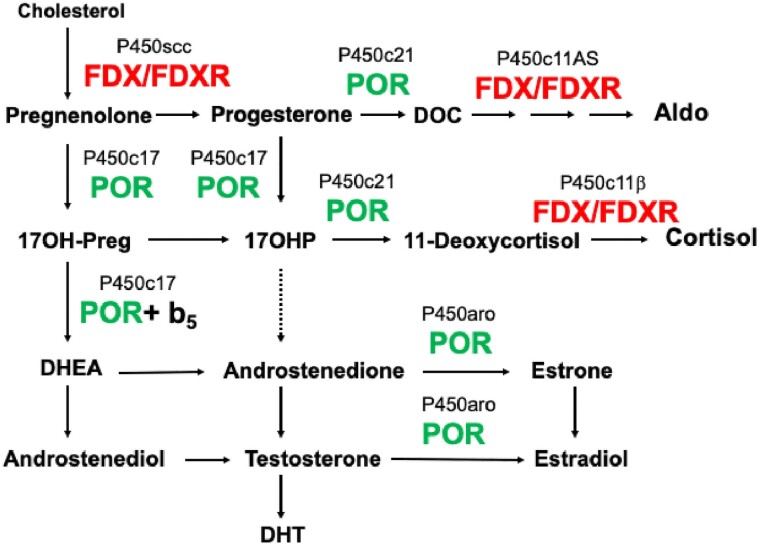
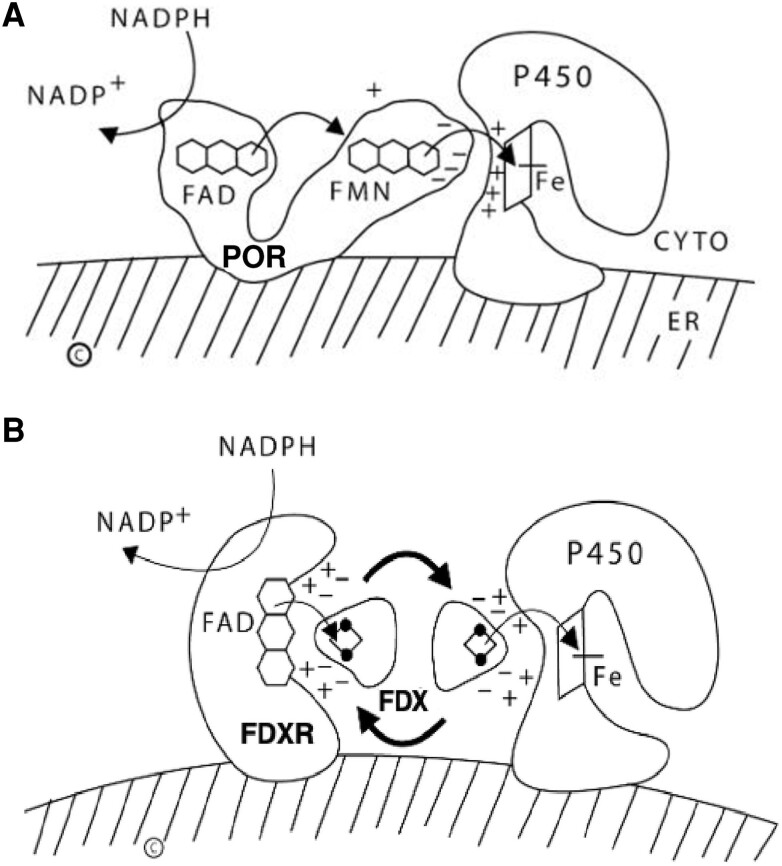
Most disorders of steroidogenesis, such as forms of congenital adrenal hyperplasia (CAH) are caused by mutations in genes encoding the steroidogenic enzymes and are often recognized clinically by cortisol deficiency, hyper- or hypo-androgenism, and/or altered mineralocorticoid function. Most steroidogenic enzymes are forms of cytochrome P450. Most P450s, including several steroidogenic enzymes, are microsomal, requiring electron donation by P450 oxidoreductase (POR); however, several steroidogenic enzymes are mitochondrial P450s, requiring electron donation via ferredoxin reductase (FDXR) and ferredoxin (FDX). POR deficiency is a rare but well-described form of CAH characterized by impaired activity of 21-hydroxylase (P450c21, CYP21A2) and 17-hydroxylase/17,20-lyase (P450c17, CYP17A1); more severely affected individuals also have the Antley-Bixler skeletal malformation syndrome and disordered genital development in both sexes, and hence is easily recognized. The 17,20-lyase activity of P450c17 requires both POR and cytochrome b5 (b5), which promote electron transfer. Mutations of POR, b5, or P450c17 can cause selective 17,20-lyase deficiency. In addition to providing electrons to mitochondrial P450s, FDX, and FDXR are required for the synthesis of iron-sulfur clusters, which are used by many enzymes. Recent work has identified FDXR mutations in patients with visual impairment, optic atrophy, neuropathic hearing loss, and developmental delay, resembling the global neurologic disorders seen with mitochondrial diseases. Many of these patients have had life-threatening events or deadly infections, often without an apparent triggering event. Adrenal insufficiency has been predicted in such individuals but has only been documented recently. Neurologists, neonatologists, and geneticists should seek endocrine assistance in evaluating and treating patients with mutations in FDXR.
期刊介绍:
The Journal of Clinical Endocrinology & Metabolism is the world"s leading peer-reviewed journal for endocrine clinical research and cutting edge clinical practice reviews. Each issue provides the latest in-depth coverage of new developments enhancing our understanding, diagnosis and treatment of endocrine and metabolic disorders. Regular features of special interest to endocrine consultants include clinical trials, clinical reviews, clinical practice guidelines, case seminars, and controversies in clinical endocrinology, as well as original reports of the most important advances in patient-oriented endocrine and metabolic research. According to the latest Thomson Reuters Journal Citation Report, JCE&M articles were cited 64,185 times in 2008.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


