{"title":"多参量方法的精确热化学:内部收缩多参量耦合簇理论的压力测试","authors":"Alexander Waigum, Murat Ertürk and Andreas Köhn*, ","doi":"10.1021/acs.jpca.4c0581910.1021/acs.jpca.4c05819","DOIUrl":null,"url":null,"abstract":"<p >The internally contracted multireference coupled-cluster method with single, double and perturbative triple excitations, icMRCCSD(T), was tested for its performance in the context of computational high-accuracy thermochemistry. The results were gauged against the standard single-reference coupled-cluster hierarchy with up to 5-fold excitations. The test set comprised of a selection of first-row dinuclear compounds and the three 3d-transition metal compounds MnH, FeH, and CoH. The results revealed two problems with the current formulation of icMRCCSD(T). First, the choice of the Dyall Hamiltonian as the zeroth-order Hamiltonian, which leads to a biased description of the different orbital subspaces and particularly poor results for the atomic correlation energies, and second, the tendency to overestimate the perturbative correction for triply excited clusters, in particular in the presence of open shells and correspondingly low orbital-energy gaps. The two problems could be solved by resorting to the effective Fock operator as zeroth-order Hamiltonian and by adopting a modified amplitude equation that includes terms quadratic in the pair clusters. A similar modification was recently proposed by Masios et al. (<i>Phys. Rev. Lett.</i> <b>2023</b>, <i>131</i>, 186401) in the context of applying single-reference coupled-cluster theory to systems with small or vanishing band gaps and we chose the acronym ‘(cT*) correction’ in analogy to that work. In contrast to the work of Masios et al., additional terms including single excitation clusters were omitted, as these again lead to an overestimation of correlation effects in more difficult cases. We also tested another alternative for the zeroth-order Hamiltonian and additional higher-order corrections for the correlation energy. These extensions did not significantly improve the results and were also computationally more demanding. The improved icMRCCSD(cT*)<sub>F</sub> method yields very accurate results with errors, relative to accurate benchmarks, better than 2 kJ/mol for total energies and atomization energies for the entire set of examples considered in this work.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 46","pages":"10053–10070 10053–10070"},"PeriodicalIF":2.7000,"publicationDate":"2024-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Accurate Thermochemistry with Multireference Methods: A Stress Test for Internally Contracted Multireference Coupled-Cluster Theory\",\"authors\":\"Alexander Waigum, Murat Ertürk and Andreas Köhn*, \",\"doi\":\"10.1021/acs.jpca.4c0581910.1021/acs.jpca.4c05819\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The internally contracted multireference coupled-cluster method with single, double and perturbative triple excitations, icMRCCSD(T), was tested for its performance in the context of computational high-accuracy thermochemistry. The results were gauged against the standard single-reference coupled-cluster hierarchy with up to 5-fold excitations. The test set comprised of a selection of first-row dinuclear compounds and the three 3d-transition metal compounds MnH, FeH, and CoH. The results revealed two problems with the current formulation of icMRCCSD(T). First, the choice of the Dyall Hamiltonian as the zeroth-order Hamiltonian, which leads to a biased description of the different orbital subspaces and particularly poor results for the atomic correlation energies, and second, the tendency to overestimate the perturbative correction for triply excited clusters, in particular in the presence of open shells and correspondingly low orbital-energy gaps. The two problems could be solved by resorting to the effective Fock operator as zeroth-order Hamiltonian and by adopting a modified amplitude equation that includes terms quadratic in the pair clusters. A similar modification was recently proposed by Masios et al. (<i>Phys. Rev. Lett.</i> <b>2023</b>, <i>131</i>, 186401) in the context of applying single-reference coupled-cluster theory to systems with small or vanishing band gaps and we chose the acronym ‘(cT*) correction’ in analogy to that work. In contrast to the work of Masios et al., additional terms including single excitation clusters were omitted, as these again lead to an overestimation of correlation effects in more difficult cases. We also tested another alternative for the zeroth-order Hamiltonian and additional higher-order corrections for the correlation energy. These extensions did not significantly improve the results and were also computationally more demanding. The improved icMRCCSD(cT*)<sub>F</sub> method yields very accurate results with errors, relative to accurate benchmarks, better than 2 kJ/mol for total energies and atomization energies for the entire set of examples considered in this work.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 46\",\"pages\":\"10053–10070 10053–10070\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-11-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c05819\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c05819","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Accurate Thermochemistry with Multireference Methods: A Stress Test for Internally Contracted Multireference Coupled-Cluster Theory
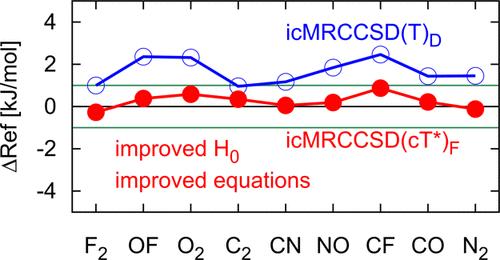
The internally contracted multireference coupled-cluster method with single, double and perturbative triple excitations, icMRCCSD(T), was tested for its performance in the context of computational high-accuracy thermochemistry. The results were gauged against the standard single-reference coupled-cluster hierarchy with up to 5-fold excitations. The test set comprised of a selection of first-row dinuclear compounds and the three 3d-transition metal compounds MnH, FeH, and CoH. The results revealed two problems with the current formulation of icMRCCSD(T). First, the choice of the Dyall Hamiltonian as the zeroth-order Hamiltonian, which leads to a biased description of the different orbital subspaces and particularly poor results for the atomic correlation energies, and second, the tendency to overestimate the perturbative correction for triply excited clusters, in particular in the presence of open shells and correspondingly low orbital-energy gaps. The two problems could be solved by resorting to the effective Fock operator as zeroth-order Hamiltonian and by adopting a modified amplitude equation that includes terms quadratic in the pair clusters. A similar modification was recently proposed by Masios et al. (Phys. Rev. Lett.2023, 131, 186401) in the context of applying single-reference coupled-cluster theory to systems with small or vanishing band gaps and we chose the acronym ‘(cT*) correction’ in analogy to that work. In contrast to the work of Masios et al., additional terms including single excitation clusters were omitted, as these again lead to an overestimation of correlation effects in more difficult cases. We also tested another alternative for the zeroth-order Hamiltonian and additional higher-order corrections for the correlation energy. These extensions did not significantly improve the results and were also computationally more demanding. The improved icMRCCSD(cT*)F method yields very accurate results with errors, relative to accurate benchmarks, better than 2 kJ/mol for total energies and atomization energies for the entire set of examples considered in this work.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


