{"title":"3T-VASP:通过多尺度梯度能量最小化实现快速非原位电化学反应器","authors":"Jonathan P. Mailoa, Xin Li, Shengyu Zhang","doi":"10.1038/s41467-024-54453-1","DOIUrl":null,"url":null,"abstract":"<p>Ab-initio methods such as density functional theory (DFT) is useful for fundamental atomistic-level study and is widely used across many scientific fields, including for the discovery of electrochemical reaction byproducts. However, many DFT steps may be needed to discover rare electrochemical reaction byproducts, which limits DFT’s scalability. In this work, we demonstrate that it is possible to generate many elementary electrochemical reaction byproducts in-silico using just a small number of ab-initio energy minimization steps if it is done in a multi-scale manner, such as via previously reported tiered tensor transform (3T) method. We first demonstrate the algorithm through a simple example of a complex floppy organic molecule passivator binding onto perovskite solar cell surface defect site. We then demonstrate more complex examples by generating hundreds of electrochemical reaction byproducts in lithium-ion battery liquid electrolyte (many are verified in previous experimental studies), with most trajectories completed within 50–100 DFT steps as opposed to more than 10,000 steps typically utilized in an ab-initio molecular dynamics trajectory. This approach requires no machine learning training data generation and can be directly applied on any new chemistries, making it suitable for ab-initio elementary chemical reaction byproduct investigation when temperature dependence is not required.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"42 1","pages":""},"PeriodicalIF":14.7000,"publicationDate":"2024-11-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"3T-VASP: fast ab-initio electrochemical reactor via multi-scale gradient energy minimization\",\"authors\":\"Jonathan P. Mailoa, Xin Li, Shengyu Zhang\",\"doi\":\"10.1038/s41467-024-54453-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Ab-initio methods such as density functional theory (DFT) is useful for fundamental atomistic-level study and is widely used across many scientific fields, including for the discovery of electrochemical reaction byproducts. However, many DFT steps may be needed to discover rare electrochemical reaction byproducts, which limits DFT’s scalability. In this work, we demonstrate that it is possible to generate many elementary electrochemical reaction byproducts in-silico using just a small number of ab-initio energy minimization steps if it is done in a multi-scale manner, such as via previously reported tiered tensor transform (3T) method. We first demonstrate the algorithm through a simple example of a complex floppy organic molecule passivator binding onto perovskite solar cell surface defect site. We then demonstrate more complex examples by generating hundreds of electrochemical reaction byproducts in lithium-ion battery liquid electrolyte (many are verified in previous experimental studies), with most trajectories completed within 50–100 DFT steps as opposed to more than 10,000 steps typically utilized in an ab-initio molecular dynamics trajectory. This approach requires no machine learning training data generation and can be directly applied on any new chemistries, making it suitable for ab-initio elementary chemical reaction byproduct investigation when temperature dependence is not required.</p>\",\"PeriodicalId\":19066,\"journal\":{\"name\":\"Nature Communications\",\"volume\":\"42 1\",\"pages\":\"\"},\"PeriodicalIF\":14.7000,\"publicationDate\":\"2024-11-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Communications\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://doi.org/10.1038/s41467-024-54453-1\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-024-54453-1","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
3T-VASP: fast ab-initio electrochemical reactor via multi-scale gradient energy minimization
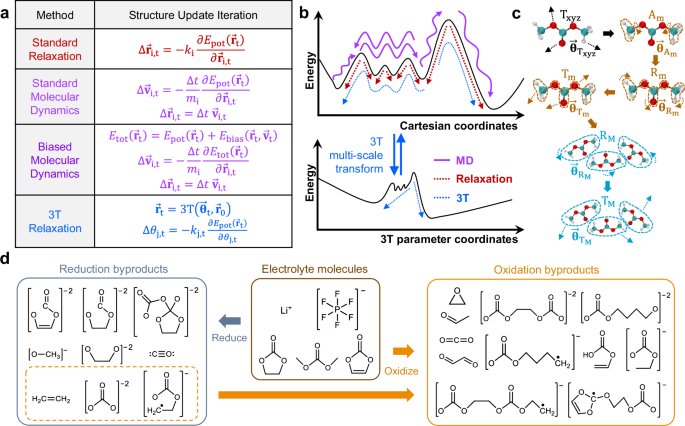
Ab-initio methods such as density functional theory (DFT) is useful for fundamental atomistic-level study and is widely used across many scientific fields, including for the discovery of electrochemical reaction byproducts. However, many DFT steps may be needed to discover rare electrochemical reaction byproducts, which limits DFT’s scalability. In this work, we demonstrate that it is possible to generate many elementary electrochemical reaction byproducts in-silico using just a small number of ab-initio energy minimization steps if it is done in a multi-scale manner, such as via previously reported tiered tensor transform (3T) method. We first demonstrate the algorithm through a simple example of a complex floppy organic molecule passivator binding onto perovskite solar cell surface defect site. We then demonstrate more complex examples by generating hundreds of electrochemical reaction byproducts in lithium-ion battery liquid electrolyte (many are verified in previous experimental studies), with most trajectories completed within 50–100 DFT steps as opposed to more than 10,000 steps typically utilized in an ab-initio molecular dynamics trajectory. This approach requires no machine learning training data generation and can be directly applied on any new chemistries, making it suitable for ab-initio elementary chemical reaction byproduct investigation when temperature dependence is not required.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


