{"title":"以孩子为原型正确诊断出患有利德尔综合征的父母和孩子:病例报告及文献综述。","authors":"Minako Tokunaga, Yuko Seki, Tatsushi Horiguchi, Kiwako Miura, Haruna Kakimoto, Satoshi Morita, Michiyo Mizota, Koshi Kusumoto, Takayasu Mori, Eisei Sohara, Shinichi Uchida, Yasuhiro Okamoto","doi":"10.1507/endocrj.EJ24-0180","DOIUrl":null,"url":null,"abstract":"<p><p>Liddle syndrome (LS) is an autosomal dominant genetic disorder characterized by early onset hypertension, hypokalemia, and low plasma aldosterone or renin concentration. It is caused by mutations in subunits of the epithelial sodium channel (ENaC). The clinical phenotypes of LS are variable and nonspecific, making it prone to both misdiagnosis and missed diagnosis. Genetic analysis is necessary to confirm the diagnosis of LS. Herein, we report the case of a 42-year-old male with LS and a 30-year history of hypertension. He was being treated for possible primary aldosteronism (PA) over the preceding 7 years; however, his hypertension was poorly controlled despite intensive combination therapy. His 13-year-old son served as a proband for a diagnosis of LS, as he had hypertension, hypokalemia, and a significant family history of hypertension. Genetic testing revealed a heterozygous pathological variant in the SCNN1B gene. This led to a diagnosis of LS, as the father was found to harbor the same mutation. Both were treated with ENaC inhibitors and a salt-restricted diet, which improved their symptoms markedly. The son's genetic diagnosis facilitated the subsequent proper diagnosis and treatment of his father. LS causes early onset hypertension; hence, its early diagnosis and treatment can prevent complications. Hereditary hypertension should be considered in cases of early onset hypertension with a significant family history. Patients diagnosed with PA using outdated criteria may have concomitant LS and require careful evaluation of biochemical and endocrine tests according to the current criteria.</p>","PeriodicalId":11631,"journal":{"name":"Endocrine journal","volume":" ","pages":"319-323"},"PeriodicalIF":2.1000,"publicationDate":"2025-03-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11913552/pdf/","citationCount":"0","resultStr":"{\"title\":\"A parent and child with Liddle syndrome diagnosed correctly with the child as the proband: a case report with review of literature.\",\"authors\":\"Minako Tokunaga, Yuko Seki, Tatsushi Horiguchi, Kiwako Miura, Haruna Kakimoto, Satoshi Morita, Michiyo Mizota, Koshi Kusumoto, Takayasu Mori, Eisei Sohara, Shinichi Uchida, Yasuhiro Okamoto\",\"doi\":\"10.1507/endocrj.EJ24-0180\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Liddle syndrome (LS) is an autosomal dominant genetic disorder characterized by early onset hypertension, hypokalemia, and low plasma aldosterone or renin concentration. It is caused by mutations in subunits of the epithelial sodium channel (ENaC). The clinical phenotypes of LS are variable and nonspecific, making it prone to both misdiagnosis and missed diagnosis. Genetic analysis is necessary to confirm the diagnosis of LS. Herein, we report the case of a 42-year-old male with LS and a 30-year history of hypertension. He was being treated for possible primary aldosteronism (PA) over the preceding 7 years; however, his hypertension was poorly controlled despite intensive combination therapy. His 13-year-old son served as a proband for a diagnosis of LS, as he had hypertension, hypokalemia, and a significant family history of hypertension. Genetic testing revealed a heterozygous pathological variant in the SCNN1B gene. This led to a diagnosis of LS, as the father was found to harbor the same mutation. Both were treated with ENaC inhibitors and a salt-restricted diet, which improved their symptoms markedly. The son's genetic diagnosis facilitated the subsequent proper diagnosis and treatment of his father. LS causes early onset hypertension; hence, its early diagnosis and treatment can prevent complications. Hereditary hypertension should be considered in cases of early onset hypertension with a significant family history. Patients diagnosed with PA using outdated criteria may have concomitant LS and require careful evaluation of biochemical and endocrine tests according to the current criteria.</p>\",\"PeriodicalId\":11631,\"journal\":{\"name\":\"Endocrine journal\",\"volume\":\" \",\"pages\":\"319-323\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2025-03-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11913552/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Endocrine journal\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1507/endocrj.EJ24-0180\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine journal","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1507/endocrj.EJ24-0180","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/21 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
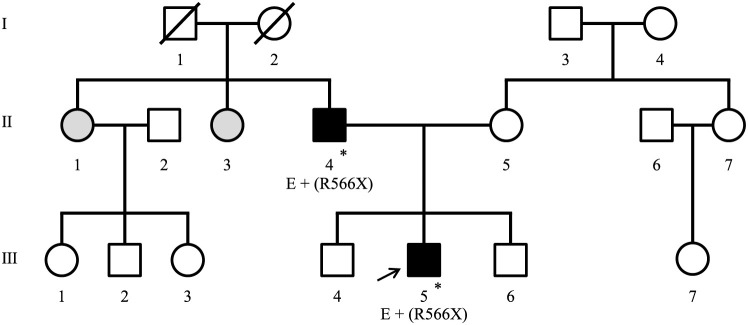
利德尔综合征(Liddle Syndrome,LS)是一种常染色体显性遗传疾病,以早发高血压、低钾血症和低血浆醛固酮或肾素浓度为特征。它是由上皮钠通道(ENaC)亚基突变引起的。LS 的临床表型多变且无特异性,因此容易误诊和漏诊。要确诊 LS,必须进行基因分析。在此,我们报告了一例 42 岁男性 LS 患者的病例,他有 30 年的高血压病史。在过去的 7 年中,他一直在接受可能是原发性醛固酮增多症(PA)的治疗;然而,尽管进行了强化综合治疗,他的高血压仍然控制不佳。他 13 岁的儿子患有高血压、低钾血症,并有重要的高血压家族史,因此可作为 LS 诊断的探查对象。基因检测显示,SCNN1B 基因存在杂合病理变异。这导致了 LS 的诊断,因为他的父亲也携带同样的基因突变。两人都接受了ENaC抑制剂和限盐饮食治疗,症状明显改善。儿子的基因诊断有助于随后对父亲进行正确的诊断和治疗。LS 会导致早发性高血压,因此早期诊断和治疗可以预防并发症。对于有明显家族史的早发性高血压患者,应考虑遗传性高血压。用过时的标准诊断为PA的患者可能同时患有LS,需要根据现行标准对生化和内分泌检查进行仔细评估。
A parent and child with Liddle syndrome diagnosed correctly with the child as the proband: a case report with review of literature.
Liddle syndrome (LS) is an autosomal dominant genetic disorder characterized by early onset hypertension, hypokalemia, and low plasma aldosterone or renin concentration. It is caused by mutations in subunits of the epithelial sodium channel (ENaC). The clinical phenotypes of LS are variable and nonspecific, making it prone to both misdiagnosis and missed diagnosis. Genetic analysis is necessary to confirm the diagnosis of LS. Herein, we report the case of a 42-year-old male with LS and a 30-year history of hypertension. He was being treated for possible primary aldosteronism (PA) over the preceding 7 years; however, his hypertension was poorly controlled despite intensive combination therapy. His 13-year-old son served as a proband for a diagnosis of LS, as he had hypertension, hypokalemia, and a significant family history of hypertension. Genetic testing revealed a heterozygous pathological variant in the SCNN1B gene. This led to a diagnosis of LS, as the father was found to harbor the same mutation. Both were treated with ENaC inhibitors and a salt-restricted diet, which improved their symptoms markedly. The son's genetic diagnosis facilitated the subsequent proper diagnosis and treatment of his father. LS causes early onset hypertension; hence, its early diagnosis and treatment can prevent complications. Hereditary hypertension should be considered in cases of early onset hypertension with a significant family history. Patients diagnosed with PA using outdated criteria may have concomitant LS and require careful evaluation of biochemical and endocrine tests according to the current criteria.
期刊介绍:
Endocrine Journal is an open access, peer-reviewed online journal with a long history. This journal publishes peer-reviewed research articles in multifaceted fields of basic, translational and clinical endocrinology. Endocrine Journal provides a chance to exchange your ideas, concepts and scientific observations in any area of recent endocrinology. Manuscripts may be submitted as Original Articles, Notes, Rapid Communications or Review Articles. We have a rapid reviewing and editorial decision system and pay a special attention to our quick, truly scientific and frequently-citable publication. Please go through the link for author guideline.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


