利用基于套接字的进程间通信加速分子动力学模拟
IF 4.8
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
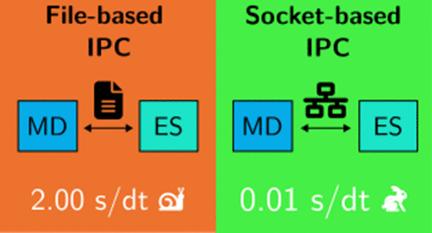
分子动力学(MD)模拟对于研究分子系统的时间演化至关重要。然而,分子动力学程序与电子结构程序之间基于文件的进程间通信(IPC)往往会制约其效率。我们提出了一种基于套接字的 IPC 实现方法,它大大加快了 MD 模拟的速度,与传统的基于文件的方法相比,计算时间缩短了 10 倍。我们的方法应用于牛顿-X 程序的非绝热分子动力学,消除了磁盘读/写开销,从而在更长的时间尺度上实现了更快的模拟。这种方法为更高效的高通量模拟打开了大门,为实时探索复杂的分子过程提供了新的机会。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Accelerating Molecular Dynamics Simulations Using Socket-Based Interprocess Communication
Molecular dynamics (MD) simulations are essential for studying the time evolution of molecular systems. Still, their efficiency is often bottlenecked by file-based interprocess communication (IPC) between MD and electronic structure programs. We present a socket-based IPC implementation that dramatically accelerates MD simulations, reducing the computational time by >10-fold compared to those of traditional file-based methods. Our approach, applied to nonadiabatic molecular dynamics with the Newton-X program, eliminates disk read/write overhead, allowing for faster simulations over longer time scales. This method opens the door to more efficient high-throughput simulations, providing new opportunities for exploring complex molecular processes in real time.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry Letters
CHEMISTRY, PHYSICAL-NANOSCIENCE & NANOTECHNOLOGY
CiteScore
9.60
自引率
7.00%
发文量
1519
审稿时长
1.6 months
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


