优化具有新型尾部杂环的 SHP2 异源抑制剂及其作为抗肿瘤治疗药物的潜力
IF 6
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
SHP2是一种与癌症有关的非受体蛋白酪氨酸磷酸酶,在许多细胞信号级联中发挥着关键作用,包括MAPK和PD-L1/PD-1途径。虽然已有几种 SHP2 异位抑制剂进入临床试验阶段,但迄今为止还没有一种获得批准。因此,迫切需要开发新的、疗效更好的 SHP2 异构抑制剂。在此,我们报告了采用基于结构的药物设计策略优化 SHP2 异构抑制剂尾部杂环的情况。我们合成了四个具有不同尾部骨架的化合物系列,其中 D13 对 SHP2 具有显著的抑制活性(IC50 = 1.2 μM)。分子对接和结合研究表明,新合成的化合物通过直接与 SHP2 结合,以相对较慢的解离速率发挥酶抑制作用。在细胞水平上,Huh7 细胞对新型 SHP2 抑制剂表现出更高的敏感性,其中 D13 通过阻滞 G0/G1 细胞周期、促进细胞凋亡和抑制 MAPK 信号通路,表现出卓越的抗增殖活性(IC50 = 38 μM)。在体内研究中,D13 在 Huh7 异种移植模型中显示出显著的抗肿瘤活性,并具有良好的可药性,口服生物利用度(F = 54%)和半衰期(t1/2 = 10.57 h)均可接受。总之,这项研究为进一步优化 SHP2 异位抑制剂的尾杂环骨架奠定了坚实的基础。本文章由计算机程序翻译,如有差异,请以英文原文为准。
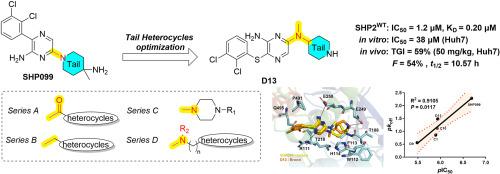
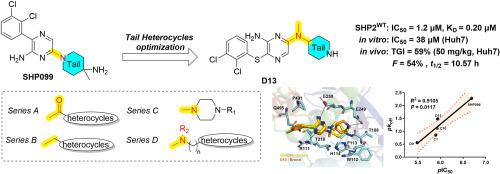
Optimization of SHP2 allosteric inhibitors with novel tail heterocycles and their potential as antitumor therapeutics
SHP2, a non-receptor protein tyrosine phosphatase involved in cancers, plays a pivotal role in numerous cellular signaling cascades, including the MAPK and PD-L1/PD-1 pathways. Although several SHP2 allosteric inhibitors have already entered clinical trials, none have been approved to date. Therefore, the development of new SHP2 allosteric inhibitors with improved efficacy is urgently required. Herein, we report the optimization of tail heterocycles in SHP2 allosteric inhibitors using a structure-based drug design strategy. Four series of compounds with different tail skeletons were synthesized, among which D13 showed notable inhibitory activity (IC50 = 1.2 μM) against SHP2. Molecular docking and binding studies indicated that the newly synthesized compounds exerted enzymatic inhibitory effects by directly binding to SHP2 with relatively slow dissociation rates. At the cellular level, Huh7 cells demonstrated heightened sensitivity to the novel SHP2 inhibitors, and D13 exhibited superior antiproliferative activity (IC50 = 38 μM) by arresting G0/G1 cell cycle, facilitating cell apoptosis and suppressing the MAPK signaling pathway. In the in vivo study, D13 displayed significant antitumor activity in a Huh7 xenograft model and possessed favorable druggability with acceptable oral bioavailability (F = 54 %) and half-life (t1/2 = 10.57 h). Collectively, this study lays a robust foundation for further optimization of the tail heterocycle skeleton in SHP2 allosteric inhibitors.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


