利用 Ab Initio 大规范蒙特卡洛对 Cl 在 Pt(111) 和 Pt(100) 上的吸附进行研究
IF 2.1
4区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
我们利用基于平面波密度泛函理论的 ab initio grand-canonical Monte Carlo (AIGCMC) 模拟来探究吸附了氯的 Pt(100) 和 Pt(111) 的结构和表面能。对于 Pt(100),我们考虑了(1 × 1)表面和(5 × 1)重构,作为实验观察到的 Pt(100) "六边形 "重构的模型。我们构建了表面能与 Cl 化学势函数的相图,并确定了最相关的表面。对于 Pt(100),我们发现六边形重构在 Cl 化学势较低时更有利,而 Cl 吸附会提升重构。预测该表面的有序结构顺序为:裸(5 × 1)Pt(100)、Θ = 1/2 (1 × 1) Pt(100)和 Θ = 2/3 (1 × 1) Pt(100),其中 Θ 是 Cl 的部分表面覆盖率。所有这些结构都可以在实验中看到。我们还在 Pt(100) 上观察到一种 Θ = 3/4 的结构以及 Pt 和 Cl 之间的混合结构,这种结构与 Θ = 2/3 时的结构有关。对于 Pt(111),我们在 Θ = 1/9、1/3、4/9、5/9 和 2/3 处发现了 (3 × 3) 单元格的递变。Θ = 1/3 和 4/9 时的结构是通过实验提出的,大多数实验预测随着 Cl 覆盖率的增加,会出现一系列 (3 × 3) 单元。如果在实验中没有发生 Cl 和 Pt 之间的混合,那么我们发现在 Θ = 1/2 处的 (4 × 2) Cl 结构在能量上更受欢迎,这与实验中观察到的情况相同。AIGCMC 的优势之一是能够在没有预定输入的情况下识别相关结构,包括无序结构。这增加了实验的高保真度和识别相关应用底物的机会。本文章由计算机程序翻译,如有差异,请以英文原文为准。
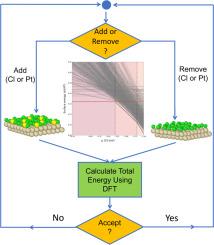
A study of Cl adsorption on Pt(111) and Pt(100) using Ab Initio Grand-canonical Monte Carlo
We used ab initio grand-canonical Monte Carlo (AIGCMC) simulations based on plane-wave density-functional theory to probe the structures and surface energies of Pt(100) and Pt(111) with adsorbed chlorine. For Pt(100), we considered both the (1 × 1) surface and a (5 × 1) reconstruction, as a model for the experimentally observed “hex” reconstruction of Pt(100). We constructed phase diagrams of the surface energies as function of the Cl chemical potential and identified the most relevant surfaces. For Pt(100), we find the hex reconstruction is favored at low Cl chemical potentials and that Cl adsorption lifts the reconstruction. The progression of ordered structures predicted for this surface is: bare (5 × 1) Pt(100), Θ = 1/2 (1 × 1) Pt(100), and Θ = 2/3 (1 × 1) Pt(100), where Θ is the fractional surface coverage of Cl. All these structures are seen experimentally. We also observe a structure with Θ = 3/4 and intermixing between Pt and Cl on Pt(100) that is related to the structure at Θ = 2/3. For Pt(111), we find a progression of (3 × 3) unit cells at Θ = 1/9, 1/3, 4/9, 5/9, and 2/3. The structures at Θ = 1/3 and 4/9 have been proposed experimentally and most experiments predict a series of (3 × 3) unit cells with increasing Cl coverage. If intermixing between Cl and Pt does not occur in experiment, then we find a (4 × 2) Cl structure at Θ = 1/2 is energetically favored, as is observed in experiment. A strength of AIGCMC is the capability to identify relevant structures, including disordered structures, without predefined input. This increases the chance of having high fidelity to experiment and identifying relevant substrates for applications.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Surface Science
化学-物理:凝聚态物理
CiteScore
3.30
自引率
5.30%
发文量
137
审稿时长
25 days
期刊介绍:
Surface Science is devoted to elucidating the fundamental aspects of chemistry and physics occurring at a wide range of surfaces and interfaces and to disseminating this knowledge fast. The journal welcomes a broad spectrum of topics, including but not limited to:
• model systems (e.g. in Ultra High Vacuum) under well-controlled reactive conditions
• nanoscale science and engineering, including manipulation of matter at the atomic/molecular scale and assembly phenomena
• reactivity of surfaces as related to various applied areas including heterogeneous catalysis, chemistry at electrified interfaces, and semiconductors functionalization
• phenomena at interfaces relevant to energy storage and conversion, and fuels production and utilization
• surface reactivity for environmental protection and pollution remediation
• interactions at surfaces of soft matter, including polymers and biomaterials.
Both experimental and theoretical work, including modeling, is within the scope of the journal. Work published in Surface Science reaches a wide readership, from chemistry and physics to biology and materials science and engineering, providing an excellent forum for cross-fertilization of ideas and broad dissemination of scientific discoveries.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


