DNTF 的热分解和冲击响应机制:深电位分子动力学模拟
IF 9
1区 工程技术
Q1 ENERGY & FUELS
引用次数: 0
摘要
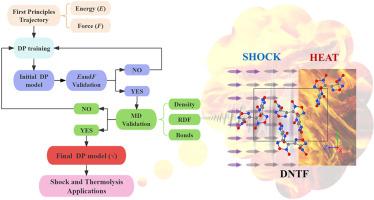
针对 3,4-双(3-硝基呋喃-4-基)呋喃(DNTF)的冲击响应和热分解机理,开发了一种神经网络深度位势(DP)。该 DP 势在 ab initio 数据集上进行训练,达到了密度泛函理论(DFT)的精度和更高的计算效率。模拟结果表明,DNTF 在冲击加载下是各向异性的。沿[100]、[010]和[001]方向的临界冲击分解温度分别为463.64 K、451.85 K和486.69 K。首次利用分子动力学结合 DP 模型成功模拟了 DNTF 的低温(750 K)分解。在热条件下,DNTF 的分解始于呋喃环的 O-N 键打开,然后是 C-NO2 键断裂和呋喃环打开。临界热分解温度为 458.3 K 至 562.5 K,加热速率为 13.5 K/ps。最终产物为 CO2、N2 和 CO,主要中间产物为 NO2、NO 和 N2O。通过小泽法得到的分解活化能为 142.4 kJ/mol。这项研究不仅为研究 DNTF 的性能提供了强有力的工具,也为其他高能材料提供了可行的方法,极大地推动了该领域的发展。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Thermal decomposition and shock response mechanism of DNTF: Deep potential molecular dynamics simulations
A neural network deep potential (DP) is developed for the shock response and thermal decomposition mechanisms of 3,4-Bis(3-nitrofurazan-4-yl)furoxan (DNTF). This DP potential, trained on ab initio datasets, achieves the accuracy of density functional theory (DFT) and higher computational efficiency. The simulation results show that DNTF is anisotropic under shock loading. And the critical shock decomposition temperatures along the [100], [010], and [001] directions are 463.64 K, 451.85 K, and 486.69 K, respectively. For the first time, the low-temperature (<750 K) decomposition of DNTF is successfully simulated using molecular dynamics combined with DP model. The decomposition of DNTF begins with the O-N bond opening of the furoxan ring, followed by the breaking of the C-NO2 bond and opening of the furazan rings, under thermal conditions. The critical thermal decomposition is between 458.3 K and 562.5 K at a heating rate of 13.5 K/ps. The final products are CO2, N2, and CO, and the main intermediates are NO2, NO, and N2O. The activation energy of decomposition is 142.4 kJ/mol obtained by Ozawa method. This study not only provides a powerful tool for investigating the performance of DNTF but also offers a feasible approach for other energetic materials, advancing the field significantly.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Energy
工程技术-能源与燃料
CiteScore
15.30
自引率
14.40%
发文量
0
审稿时长
14.2 weeks
期刊介绍:
Energy is a multidisciplinary, international journal that publishes research and analysis in the field of energy engineering. Our aim is to become a leading peer-reviewed platform and a trusted source of information for energy-related topics.
The journal covers a range of areas including mechanical engineering, thermal sciences, and energy analysis. We are particularly interested in research on energy modelling, prediction, integrated energy systems, planning, and management.
Additionally, we welcome papers on energy conservation, efficiency, biomass and bioenergy, renewable energy, electricity supply and demand, energy storage, buildings, and economic and policy issues. These topics should align with our broader multidisciplinary focus.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


