2-thiophenecarbonitrile 的抗炎和抗氧化活性、毒性预测、计算调查和分子对接研究
IF 3.6
3区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
摘要
密度泛函理论(DFT)和分子对接是现代化学和药物设计中举足轻重的计算技术。本研究探讨了 2-噻吩甲腈(2TCN)的电子结构和反应性,重点研究了 HOMO-LUMO 能隙、MEP、Mulliken 原子电荷、自然种群分析和 Mutiwfn(ELF、LOL、ALIE 和 RDG)分析等重要因素。在乙腈、水、气体和甲醇等不同溶剂中计算了 MEP 和 FMO。抗炎和抗氧化研究表明,2TCN 具有很强的活性。此外,还进行了分子对接研究,以阐明化合物与靶蛋白之间的结合相互作用,从而深入了解其潜在的治疗机制。结果显示了结合能、相互作用残基和最有利的对接姿势。这种方法强调了理论和计算方法在推进分子设计和治疗发现方面的结合。本文章由计算机程序翻译,如有差异,请以英文原文为准。
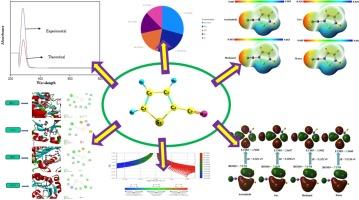
Anti-inflammatory and antioxidant activity, toxicity prediction, computational investigation, and molecular docking studies of 2-thiophenecarbonitrile
Density Functional Theory (DFT) and Molecular docking are pivotal computational techniques in modern chemistry and drug design. This work investigates the electronic structure and reactivity of 2-thiophenecarbonitrile (2TCN) with an emphasis on important factors such as HOMO-LUMO energy gap, MEP, Mulliken atomic charges, natural population analysis, and Mutiwfn (ELF, LOL, ALIE, and RDG) analysis. The MEP and FMO studies were calculated in various solvents like acetonitrile, water, gas, and methanol. The anti-inflammatory and antioxidant investigations revealed substantial activities by 2TCN. Additionally, molecular docking studies are performed to elucidate the binding interaction between the compound and target proteins, providing insights into its potential therapeutic mechanisms. The results demonstrate the binding energies, interaction residues, and the most favorable docking poses. This approach underscores the integration of theoretical and computational methods in advancing molecule design and therapeutic discovery.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of King Saud University - Science
Multidisciplinary-Multidisciplinary
CiteScore
7.20
自引率
2.60%
发文量
642
审稿时长
49 days
期刊介绍:
Journal of King Saud University – Science is an official refereed publication of King Saud University and the publishing services is provided by Elsevier. It publishes peer-reviewed research articles in the fields of physics, astronomy, mathematics, statistics, chemistry, biochemistry, earth sciences, life and environmental sciences on the basis of scientific originality and interdisciplinary interest. It is devoted primarily to research papers but short communications, reviews and book reviews are also included. The editorial board and associated editors, composed of prominent scientists from around the world, are representative of the disciplines covered by the journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


