机器学习辅助筛选氮掺杂石墨烯基双原子氢进化电催化剂
IF 3.9
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
氮掺杂石墨烯基双原子催化剂(NG-DACs)通常在氢气进化反应(HER)中表现出较高的反应活性。然而,基于 DACs 的高效 HER 催化剂的实验设计既耗时又昂贵。本研究采用密度泛函理论结合机器学习(DFT-ML)的策略来快速筛选 NG-DACs HER 催化剂。通过 DFT 计算获得的 HER 中间体在 NG-DACs 上的吸附能及其元素特征被用于构建 ML 数据库。评估了三种 ML 算法的预测性能,发现梯度提升回归(GBR)的预测精度最高。以 Pt (111) 上的 HER 能量为参考,快速筛选出了具有高 HER 活性的 NG-DAC,最终确定了 24 种潜在催化剂。通过氢吸附吉布斯自由能分析,进一步筛选和验证了 ML 筛选出的催化剂。因此,利用所提出的基于 DFT-ML 的催化剂预测框架,预测出了四种有前景的 NG-DAC HER 催化剂。DFT-ML 策略为筛选新型 HER 电催化剂提供了一种很有前景的方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
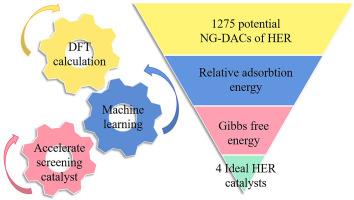
Machine learning assisted screening of nitrogen-doped graphene-based dual-atom hydrogen evolution electrocatalysts
Nitrogen doped graphene-based dual-atom catalysts (NG-DACs) usually exhibit high reactivity for hydrogen evolution reaction (HER). The experimental design of efficient DACs-based HER catalysts, however, is time-consuming and expensive. In this work, a density functional theory combined with machine learning (DFT-ML) strategy is adopted towards the rapid screening of NG-DACs HER catalyst. The adsorption energies of HER intermediates on NG-DACs obtained by DFT calculations and their elemental features were used for the ML database construction. The prediction performance of three ML algorithms was evaluated, and gradient boosted regression (GBR) is found exhibit the best prediction accuracy. Using the HER energetics on Pt (111) as reference, quick screening of the NG-DACs with high HER activity was achieved, resulting to twenty-four potential catalysts. Further selection and validation of the ML-screened catalysts were performed by the hydrogen adsorption Gibbs free energy analysis. Accordingly, four promising NG-DACs HER catalysts were predicted using the proposed DFT-ML based catalyst prediction framework. The DFT-ML strategy offers an promising approach for the screening of new HER electrocatalysts.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Molecular Catalysis
Chemical Engineering-Process Chemistry and Technology
CiteScore
6.90
自引率
10.90%
发文量
700
审稿时长
40 days
期刊介绍:
Molecular Catalysis publishes full papers that are original, rigorous, and scholarly contributions examining the molecular and atomic aspects of catalytic activation and reaction mechanisms. The fields covered are:
Heterogeneous catalysis including immobilized molecular catalysts
Homogeneous catalysis including organocatalysis, organometallic catalysis and biocatalysis
Photo- and electrochemistry
Theoretical aspects of catalysis analyzed by computational methods
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


