适当的数控配位提高了石墨烯边缘锚定铂单原子将甲烷和二氧化碳转化为醋酸的催化活性
IF 3.9
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
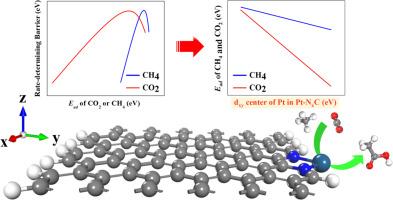
将 CH4 和 CO2 直接转化为醋酸的反应在化学工业中有着广泛而重要的应用。在这项工作中,我们基于密度泛函理论(DFT)计算,对锚定在掺杂 N 的石墨烯边缘的单铂原子催化剂的催化性能进行了系统的计算化学研究。DFT 计算结果表明,单个铂原子的催化活性受局部 N 原子配位的影响很大。微动模型显示,在 600 K 和 2 bar 条件下,CH3COOH 在 Pt-N1C 催化剂上的 TOF 达到 7.63×102 s-1 进一步分析表明,反应物 CH4 和 CO2 的吸附强度与铂原子 dxy 轨道中心的能级成线性关系。Pt-N1C 对 CH4 和 CO2 的吸附强度适中,因此在转化反应过程中甲烷更容易活化,H 和 CH3 更容易迁移。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Proper NCoordination improves catalytic activity of graphene edge anchored Pt single atom for conversion of methane and carbon dioxide to acetic acid
The reaction of directly converting CH4 and CO2 into acetic acid has a wide and important application in the chemical industry. In this work, we carried out systematically computational chemistry study on the catalytic performance of the single Pt atom catalyst anchored at the edge of N-doped graphene for the direct co-conversion of CH4 and CO2 to acetic acid based on density functional theory (DFT) calculations. The DFT calculation results show the catalytic activity of single Pt atom is significantly tuned by the local N-atom coordination. The Pt-N1C exhibits the best catalytic performance of CH4 and CO2 conversion with a low rate-determining free energy barrier of 0.69 eV The microkinetic modeling shows that the TOF of CH3COOH on Pt-N1C catalyst reaches 7.63×102 s−1 at 600 K and 2 bar Further analysis shows that the adsorption strength of reactant CH4 and CO2 is linearly correlated with the energy level of dxy orbital center of Pt atom. A moderate adsorption strength of CH4 and CO2 over the Pt-N1C leads to easier activation of methane and migration of H and CH3 during the conversion reaction.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Molecular Catalysis
Chemical Engineering-Process Chemistry and Technology
CiteScore
6.90
自引率
10.90%
发文量
700
审稿时长
40 days
期刊介绍:
Molecular Catalysis publishes full papers that are original, rigorous, and scholarly contributions examining the molecular and atomic aspects of catalytic activation and reaction mechanisms. The fields covered are:
Heterogeneous catalysis including immobilized molecular catalysts
Homogeneous catalysis including organocatalysis, organometallic catalysis and biocatalysis
Photo- and electrochemistry
Theoretical aspects of catalysis analyzed by computational methods
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


