双功能聚氧化金属酸盐团簇上氧化还原和布氏位点的热化学相关性及其在甲醇-O2 催化中的动力学后果
IF 11.3
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
甲醇氧化脱氢和脱水的动力学相互关联性体现了双功能 Keggin 型多氧金属硫酸盐(POM)磷钼酸簇上氧化还原位点和布氏位点之间的潜在热化学/电子学相关性,其电子特性受到钠离子交换(HxNa3-xPMo,x = 3-0)的干扰。随着钠交换量的增加,在 433 K 时,甲醇氧化脱氢过程中发生在孤立氧化还原位点(O*)或布氏酸-氧化还原位点对(OH/O*)上的基本 C-H 裂解的活化自由能和甲醇脱水过程中发生在布氏位点上的一阶 C-O 形成的活化自由能在 10-11 kJ mol-1 的范围内成正比增加,而它们的活化焓则呈现出反相关性。博恩-哈伯热化学分析通过建立它们各自的动力学-热化学关系,揭示了这些位点相互关联性背后的原因。与动力学相关的 C-H 裂解涉及一个晚期过渡态,即 O* 处的 [HOCH2---H---O*]‡ 或 OH/O* 处的 [OH--HOCH2--H--O*]‡,其中一个电子 (e-) 和一个质子 (H+) 作为一个 H 原子 (H-) 从甲基片段转移到氧化还原位点、其中氢加成能(HAE)由 POM 簇的负电子亲和力(-EAPOM)和质子亲和力(-PA)组成,是一个动力学描述因子。并联甲醇 C-O 形成的特点是后期碳化转变态,即[(CH3OH---CH3+---H2O)---POM-]‡,涉及质子从 POM 团簇转移到吸附的甲醇物种,其中布氏位点的去质子化能 (DPE) 是动力学描述因子。值得注意的是,随着钠交换量的增加,氢加成能减少了 ∼23 kJ mol-1,而去质子化能增加了 80-230 kJ mol-1。HxNa3-xPMo 团簇(x = 3-1)上钠交换时,电子转移的能量效应(-EAPOM)调节了氧化还原(-PA)和布氏酸催化(DPE)过程中质子转移的固有对立性,从而产生了这种轻微的热化学负相关。本文所建立的机理解释和框架明确地将氧化还原位点和布伦斯特位点的动力学、热化学和电子特性联系起来,深入揭示了它们内在的反应性耦合,并适用于其他双功能催化剂。本文章由计算机程序翻译,如有差异,请以英文原文为准。
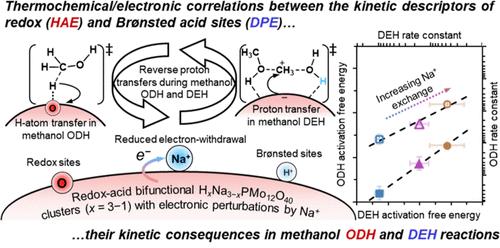
Thermochemical Correlations of Redox and Brønsted Sites on Bifunctional Polyoxometalate Clusters and Their Kinetic Consequences in Methanol-O2 Catalysis
Kinetic interconnectivities of methanol oxidative dehydrogenation and dehydration are manifestation of the underlying thermochemical/electronic correlations between redox and Brønsted sites on bifunctional Keggin-type polyoxometalate (POM) phosphomolybdic acid clusters with their electronic properties perturbed by sodium cation exchange (HxNa3–xPMo, x = 3–0). As sodium exchange increases, activation free energies for the elementary C–H scission in methanol oxidative dehydrogenation, occurring at isolated redox sites (O*) or Brønsted acid-redox site pairs (OH/O*), and for the first-order C–O formation in methanol dehydration, occurring at Brønsted sites, increase proportionally within 10–11 kJ mol–1 at 433 K, while their activation enthalpies exhibit an inverse correlation. A Born–Haber thermochemical analysis reveals the reasons behind the site interconnectivities by establishing their respective kinetic-thermochemical relationships. The kinetically relevant C–H scission involves a late transition state, either [HOCH2···H···O*]‡ at O* or [OH···HOCH2···H···O*]‡ at OH/O*, with the transfer of an electron (e–) and a proton (H+) as an H atom (H•) from the methyl fragment to redox sites, where hydrogen addition energy (HAE), comprising the negative electron affinity (−EAPOM) and proton affinity (−PA) of POM clusters, is a kinetic descriptor. The parallel methanol C–O formation features a late carbocationic transition state, [(CH3OH···CH3+···H2O)···POM–]‡, involving proton transfer from POM clusters to adsorbed methanol species, where the deprotonation energy (DPE) of the Brønsted site serves as a kinetic descriptor. Notably, hydrogen addition energy decreases by ∼23 kJ mol–1, while deprotonation energy increases by 80–230 kJ mol–1, as sodium exchange increases. This slight negative thermochemical correlation arises from the inherent opposing proton transfers during redox (−PA) and Brønsted acid catalysis (DPE), modulated by the energetic effect of electron transfer (−EAPOM) upon sodium exchange on HxNa3–xPMo clusters (x = 3–1). The mechanistic interpretation and framework established here explicitly correlate the kinetic, thermochemical, and electronic properties of redox and Brønsted sites, offering insights into their intrinsic reactivity couplings, and are applicable to other bifunctional catalysts.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


