Nader M. Boshta, Michael Lewash, Meryem Köse, Vigneshwaran Namasivayam, Soumya Sarkar, Jan H. Voss, Andy J. Liedtke, Anna Junker, Maoqun Tian, Anne Stößel, Mahmoud Rashed, Ahmed Mahal, Nicole Merten, Cécile Pegurier, Jörg Hockemeyer, Evi Kostenis and Christa E. Müller*,
{"title":"发现可拮抗促炎孤儿 G 蛋白偶联受体 GPR17 的蒽酸衍生物","authors":"Nader M. Boshta, Michael Lewash, Meryem Köse, Vigneshwaran Namasivayam, Soumya Sarkar, Jan H. Voss, Andy J. Liedtke, Anna Junker, Maoqun Tian, Anne Stößel, Mahmoud Rashed, Ahmed Mahal, Nicole Merten, Cécile Pegurier, Jörg Hockemeyer, Evi Kostenis and Christa E. Müller*, ","doi":"10.1021/acs.jmedchem.4c0175510.1021/acs.jmedchem.4c01755","DOIUrl":null,"url":null,"abstract":"<p >The G protein-coupled receptor 17 (GPR17) is an orphan receptor involved in inflammatory diseases. GPR17 antagonists have been proposed for the treatment of multiple sclerosis due to their potential to induce remyelination. Potent, selective antagonists are required to enable target validation. In the present study, we describe the discovery of a novel class of GPR17 antagonists based on an anthranilic acid scaffold. The compounds’ potencies were evaluated in calcium mobilization and radioligand binding assays, and structure–activity relationships were analyzed. Selected antagonists were additionally studied in cAMP and G protein activation assays. The most potent antagonists were 5-methoxy-2-(5-(3′-methoxy-[1,1′-biphenyl]-2-yl)furan-2-carboxamido)benzoic acid (<b>52</b>, PSB-22269, K<sub>i</sub> 8.91 nM) and its 3′-trifluoromethyl analog (<b>54</b>, PSB-24040, K<sub>i</sub> 83.2 nM). Receptor–ligand docking studies revealed that the compounds’ binding site is characterized by positively charged arginine residues and a lipophilic pocket. These findings yield valuable insights into this poorly characterized receptor providing a basis for future drug development.</p>","PeriodicalId":46,"journal":{"name":"Journal of Medicinal Chemistry","volume":"67 21","pages":"19365–19394 19365–19394"},"PeriodicalIF":6.8000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Discovery of Anthranilic Acid Derivatives as Antagonists of the Pro-Inflammatory Orphan G Protein-Coupled Receptor GPR17\",\"authors\":\"Nader M. Boshta, Michael Lewash, Meryem Köse, Vigneshwaran Namasivayam, Soumya Sarkar, Jan H. Voss, Andy J. Liedtke, Anna Junker, Maoqun Tian, Anne Stößel, Mahmoud Rashed, Ahmed Mahal, Nicole Merten, Cécile Pegurier, Jörg Hockemeyer, Evi Kostenis and Christa E. Müller*, \",\"doi\":\"10.1021/acs.jmedchem.4c0175510.1021/acs.jmedchem.4c01755\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The G protein-coupled receptor 17 (GPR17) is an orphan receptor involved in inflammatory diseases. GPR17 antagonists have been proposed for the treatment of multiple sclerosis due to their potential to induce remyelination. Potent, selective antagonists are required to enable target validation. In the present study, we describe the discovery of a novel class of GPR17 antagonists based on an anthranilic acid scaffold. The compounds’ potencies were evaluated in calcium mobilization and radioligand binding assays, and structure–activity relationships were analyzed. Selected antagonists were additionally studied in cAMP and G protein activation assays. The most potent antagonists were 5-methoxy-2-(5-(3′-methoxy-[1,1′-biphenyl]-2-yl)furan-2-carboxamido)benzoic acid (<b>52</b>, PSB-22269, K<sub>i</sub> 8.91 nM) and its 3′-trifluoromethyl analog (<b>54</b>, PSB-24040, K<sub>i</sub> 83.2 nM). Receptor–ligand docking studies revealed that the compounds’ binding site is characterized by positively charged arginine residues and a lipophilic pocket. These findings yield valuable insights into this poorly characterized receptor providing a basis for future drug development.</p>\",\"PeriodicalId\":46,\"journal\":{\"name\":\"Journal of Medicinal Chemistry\",\"volume\":\"67 21\",\"pages\":\"19365–19394 19365–19394\"},\"PeriodicalIF\":6.8000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Medicinal Chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01755\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01755","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
摘要
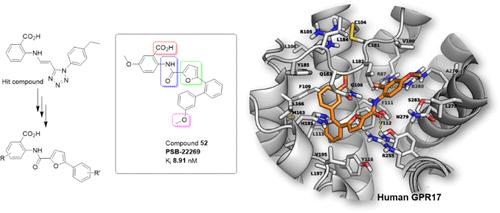
G 蛋白偶联受体 17(GPR17)是一种参与炎症性疾病的孤儿受体。GPR17 拮抗剂具有诱导再髓鞘化的潜力,因此被提议用于治疗多发性硬化症。需要强效、选择性的拮抗剂来实现目标验证。在本研究中,我们介绍了基于蒽酸支架发现的一类新型 GPR17 拮抗剂。我们在钙动员和放射性配体结合试验中评估了这些化合物的效力,并分析了它们之间的结构-活性关系。此外,还在 cAMP 和 G 蛋白活化试验中对选定的拮抗剂进行了研究。最有效的拮抗剂是 5-甲氧基-2-(5-(3′-甲氧基-[1,1′-联苯]-2-基)呋喃-2-甲酰胺基)苯甲酸(52,PSB-22269,Ki 8.91 nM)及其 3′-三氟甲基类似物(54,PSB-24040,Ki 83.2 nM)。受体-配体对接研究显示,这些化合物的结合位点以带正电荷的精氨酸残基和一个亲脂口袋为特征。这些发现为今后的药物开发提供了基础。
Discovery of Anthranilic Acid Derivatives as Antagonists of the Pro-Inflammatory Orphan G Protein-Coupled Receptor GPR17
The G protein-coupled receptor 17 (GPR17) is an orphan receptor involved in inflammatory diseases. GPR17 antagonists have been proposed for the treatment of multiple sclerosis due to their potential to induce remyelination. Potent, selective antagonists are required to enable target validation. In the present study, we describe the discovery of a novel class of GPR17 antagonists based on an anthranilic acid scaffold. The compounds’ potencies were evaluated in calcium mobilization and radioligand binding assays, and structure–activity relationships were analyzed. Selected antagonists were additionally studied in cAMP and G protein activation assays. The most potent antagonists were 5-methoxy-2-(5-(3′-methoxy-[1,1′-biphenyl]-2-yl)furan-2-carboxamido)benzoic acid (52, PSB-22269, Ki 8.91 nM) and its 3′-trifluoromethyl analog (54, PSB-24040, Ki 83.2 nM). Receptor–ligand docking studies revealed that the compounds’ binding site is characterized by positively charged arginine residues and a lipophilic pocket. These findings yield valuable insights into this poorly characterized receptor providing a basis for future drug development.
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


