Dongyu Liu, Bayan Amer Abzakh, Elena A. Kazakova, Dmitrii A. Abrameshin, Pavel A. Troshin, Run Long and Andrey S. Vasenko*,
{"title":"波函数局部化降低了无序双包晶石 Cs2AgBiBr6 的带隙","authors":"Dongyu Liu, Bayan Amer Abzakh, Elena A. Kazakova, Dmitrii A. Abrameshin, Pavel A. Troshin, Run Long and Andrey S. Vasenko*, ","doi":"10.1021/acs.jpclett.4c0294610.1021/acs.jpclett.4c02946","DOIUrl":null,"url":null,"abstract":"<p >Double perovskite Cs<sub>2</sub>AgBiBr<sub>6</sub> is a promising alternative to lead-based perovskites with excellent stability and attractive optoelectronic properties. However, a relatively large bandgap severely limits its performance in many applications such as solar cells and photodetectors. It has been reported that a random distribution of Ag and Bi atoms in Cs<sub>2</sub>AgBiBr<sub>6</sub> effectively reduces its bandgap without introducing dopants or impurities, while the mechanism remains unclear. Here, using density functional theory calculations, we demonstrate that the Ag–Bi disorder in Cs<sub>2</sub>AgBiBr<sub>6</sub> generates localized electronic states as band edges to regulate the bandgap. The disordered structures segregate Ag and Bi atoms in the lattice, and the formed homoatomic clusters lead to wave function localization. Moreover, the bandgap decrease exhibits a non-monotonic dependence on the degree of disorder. Our results are comparable with experimental observations and provide crucial insights into understanding the order–disorder transition in double perovskites.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"15 45","pages":"11268–11274 11268–11274"},"PeriodicalIF":4.8000,"publicationDate":"2024-11-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Wave Function Localization Reduces the Bandgap of Disordered Double Perovskite Cs2AgBiBr6\",\"authors\":\"Dongyu Liu, Bayan Amer Abzakh, Elena A. Kazakova, Dmitrii A. Abrameshin, Pavel A. Troshin, Run Long and Andrey S. Vasenko*, \",\"doi\":\"10.1021/acs.jpclett.4c0294610.1021/acs.jpclett.4c02946\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Double perovskite Cs<sub>2</sub>AgBiBr<sub>6</sub> is a promising alternative to lead-based perovskites with excellent stability and attractive optoelectronic properties. However, a relatively large bandgap severely limits its performance in many applications such as solar cells and photodetectors. It has been reported that a random distribution of Ag and Bi atoms in Cs<sub>2</sub>AgBiBr<sub>6</sub> effectively reduces its bandgap without introducing dopants or impurities, while the mechanism remains unclear. Here, using density functional theory calculations, we demonstrate that the Ag–Bi disorder in Cs<sub>2</sub>AgBiBr<sub>6</sub> generates localized electronic states as band edges to regulate the bandgap. The disordered structures segregate Ag and Bi atoms in the lattice, and the formed homoatomic clusters lead to wave function localization. Moreover, the bandgap decrease exhibits a non-monotonic dependence on the degree of disorder. Our results are comparable with experimental observations and provide crucial insights into understanding the order–disorder transition in double perovskites.</p>\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"15 45\",\"pages\":\"11268–11274 11268–11274\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-11-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpclett.4c02946\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.4c02946","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
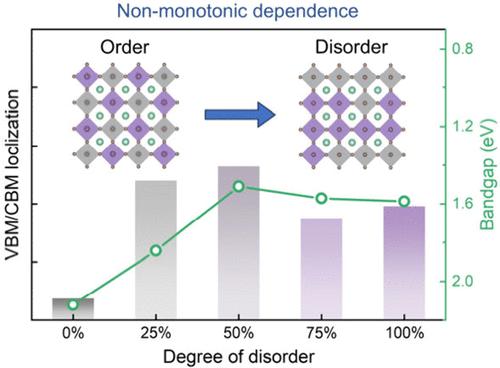
双包晶石 Cs2AgBiBr6 具有出色的稳定性和诱人的光电特性,是铅基包晶石的理想替代品。然而,相对较大的带隙严重限制了它在太阳能电池和光电探测器等许多应用中的性能。据报道,Cs2AgBiBr6 中银原子和铋原子的随机分布可在不引入掺杂剂或杂质的情况下有效降低其带隙,但其机理仍不清楚。在这里,我们利用密度泛函理论计算证明,Cs2AgBiBr6 中的银铋无序结构会产生局部电子态,作为带边来调节带隙。无序结构在晶格中分离了 Ag 原子和 Bi 原子,形成的同原子团簇导致了波函数的局部化。此外,带隙的减小与无序程度呈非单调依赖关系。我们的研究结果与实验观察结果相当,为理解双包晶的有序-无序转变提供了重要见解。
Wave Function Localization Reduces the Bandgap of Disordered Double Perovskite Cs2AgBiBr6
Double perovskite Cs2AgBiBr6 is a promising alternative to lead-based perovskites with excellent stability and attractive optoelectronic properties. However, a relatively large bandgap severely limits its performance in many applications such as solar cells and photodetectors. It has been reported that a random distribution of Ag and Bi atoms in Cs2AgBiBr6 effectively reduces its bandgap without introducing dopants or impurities, while the mechanism remains unclear. Here, using density functional theory calculations, we demonstrate that the Ag–Bi disorder in Cs2AgBiBr6 generates localized electronic states as band edges to regulate the bandgap. The disordered structures segregate Ag and Bi atoms in the lattice, and the formed homoatomic clusters lead to wave function localization. Moreover, the bandgap decrease exhibits a non-monotonic dependence on the degree of disorder. Our results are comparable with experimental observations and provide crucial insights into understanding the order–disorder transition in double perovskites.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


