Nickolas A. Joyner, João Gabriel Farias Romeu, Cole R. Durkee and David A. Dixon*,
{"title":"二原子硫化镍的电子结构","authors":"Nickolas A. Joyner, João Gabriel Farias Romeu, Cole R. Durkee and David A. Dixon*, ","doi":"10.1021/acs.jpca.4c0635610.1021/acs.jpca.4c06356","DOIUrl":null,"url":null,"abstract":"<p >The nature of the Ni–S bond is investigated due to its role in the absorption of atmospheric Lewis acid gases such as SO<sub>2</sub> and SO<sub>3</sub> onto Ni surfaces. The vibrational frequency and electronic structure of NiS were predicted using CCSD(T), CASSCF, and internally contracted multireference configuration interaction (icMRCI) + Q. 43 density functional theory (DFT) functionals were benchmarked. CASSCF predicted the ground state of NiS to be the <sup>5</sup>Δ state arising from the 3d<sup>8</sup>(<sup>3</sup>F)4s<sup>2</sup> (<sup>3</sup>F) and 3d<sup>9</sup>(<sup>2</sup>D)4s (<sup>3</sup>D) electronic configurations of Ni. When dynamical correlation effects are included at the icMRCI + Q level, the ground state of Ni–S is predicted to be <sup>3</sup>Σ<sup>–</sup> consistent with the experiment. The vibrational frequency of Ni–S is calculated to be 519.1 cm<sup>–1</sup> at the icMRCI + Q level, in reasonable agreement with the experimental value of 512.68 cm<sup>–1</sup>. CCSD(T) predicts the frequency of Ni–S to be 543.2 cm<sup>–1</sup> when extrapolated to the complete basis set (CBS) limit. The Feller–Peterson–Dixon value based on the CCSD(T)/CBS extrapolation for the bond dissociation energy of NiS is 350.6 kJ/mol, within <4 kJ/mol of experiment. Of the 43 DFT functionals, BP86 and O3LYP predicted the vibrational frequency in closest agreement with the experiment. The applicability of DFT to such acid gas systems was further demonstrated by calculating the energy for displacement of NiO by SO to yield NiS and O<sub>2</sub>. This displacement energy was calculated to be within experimental error for ∼50% of the DFT functionals, but large differences were also predicted for some functionals.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 45","pages":"9771–9781 9771–9781"},"PeriodicalIF":2.7000,"publicationDate":"2024-11-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Electronic Structure of Diatomic Nickel Sulfide\",\"authors\":\"Nickolas A. Joyner, João Gabriel Farias Romeu, Cole R. Durkee and David A. Dixon*, \",\"doi\":\"10.1021/acs.jpca.4c0635610.1021/acs.jpca.4c06356\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The nature of the Ni–S bond is investigated due to its role in the absorption of atmospheric Lewis acid gases such as SO<sub>2</sub> and SO<sub>3</sub> onto Ni surfaces. The vibrational frequency and electronic structure of NiS were predicted using CCSD(T), CASSCF, and internally contracted multireference configuration interaction (icMRCI) + Q. 43 density functional theory (DFT) functionals were benchmarked. CASSCF predicted the ground state of NiS to be the <sup>5</sup>Δ state arising from the 3d<sup>8</sup>(<sup>3</sup>F)4s<sup>2</sup> (<sup>3</sup>F) and 3d<sup>9</sup>(<sup>2</sup>D)4s (<sup>3</sup>D) electronic configurations of Ni. When dynamical correlation effects are included at the icMRCI + Q level, the ground state of Ni–S is predicted to be <sup>3</sup>Σ<sup>–</sup> consistent with the experiment. The vibrational frequency of Ni–S is calculated to be 519.1 cm<sup>–1</sup> at the icMRCI + Q level, in reasonable agreement with the experimental value of 512.68 cm<sup>–1</sup>. CCSD(T) predicts the frequency of Ni–S to be 543.2 cm<sup>–1</sup> when extrapolated to the complete basis set (CBS) limit. The Feller–Peterson–Dixon value based on the CCSD(T)/CBS extrapolation for the bond dissociation energy of NiS is 350.6 kJ/mol, within <4 kJ/mol of experiment. Of the 43 DFT functionals, BP86 and O3LYP predicted the vibrational frequency in closest agreement with the experiment. The applicability of DFT to such acid gas systems was further demonstrated by calculating the energy for displacement of NiO by SO to yield NiS and O<sub>2</sub>. This displacement energy was calculated to be within experimental error for ∼50% of the DFT functionals, but large differences were also predicted for some functionals.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 45\",\"pages\":\"9771–9781 9771–9781\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-11-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c06356\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c06356","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
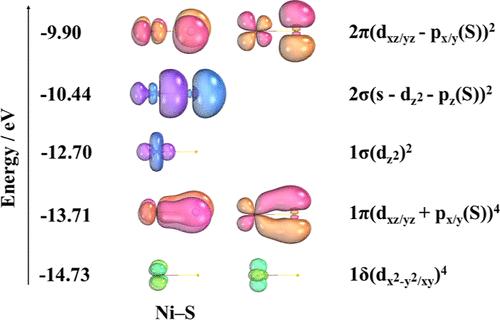
The nature of the Ni–S bond is investigated due to its role in the absorption of atmospheric Lewis acid gases such as SO2 and SO3 onto Ni surfaces. The vibrational frequency and electronic structure of NiS were predicted using CCSD(T), CASSCF, and internally contracted multireference configuration interaction (icMRCI) + Q. 43 density functional theory (DFT) functionals were benchmarked. CASSCF predicted the ground state of NiS to be the 5Δ state arising from the 3d8(3F)4s2 (3F) and 3d9(2D)4s (3D) electronic configurations of Ni. When dynamical correlation effects are included at the icMRCI + Q level, the ground state of Ni–S is predicted to be 3Σ– consistent with the experiment. The vibrational frequency of Ni–S is calculated to be 519.1 cm–1 at the icMRCI + Q level, in reasonable agreement with the experimental value of 512.68 cm–1. CCSD(T) predicts the frequency of Ni–S to be 543.2 cm–1 when extrapolated to the complete basis set (CBS) limit. The Feller–Peterson–Dixon value based on the CCSD(T)/CBS extrapolation for the bond dissociation energy of NiS is 350.6 kJ/mol, within <4 kJ/mol of experiment. Of the 43 DFT functionals, BP86 and O3LYP predicted the vibrational frequency in closest agreement with the experiment. The applicability of DFT to such acid gas systems was further demonstrated by calculating the energy for displacement of NiO by SO to yield NiS and O2. This displacement energy was calculated to be within experimental error for ∼50% of the DFT functionals, but large differences were also predicted for some functionals.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


