{"title":"静电相互作用在二甲苯 HER 动力学中被忽视的作用:超越传统描述符 ΔG ∼ 0,识别真正的活性位点","authors":"Xiang Huang, Xiangting Hu, Jiong Wang and Hu Xu*, ","doi":"10.1021/acs.jpclett.4c0258810.1021/acs.jpclett.4c02588","DOIUrl":null,"url":null,"abstract":"<p >Understanding the atomic-level mechanism of the hydrogen evolution reaction (HER) on MXene materials is crucial for developing affordable HER catalysts, while their complex surface terminations present a substantial challenge. Herein, employing constant-potential grand canonical density functional theory calculations, we elucidate the reaction kinetics of the HER on MXenes with various surface terminations by taking experimentally reported Mo<sub>2</sub>C as a prototype. We observe a contradictory scenario on Mo<sub>2</sub>C MXene when using conventional thermodynamic descriptor Δ<i>G</i><sub>H*</sub> (hydrogen binding energy). Both competing surface phases that emerge close to the equilibrium potential meet the Δ<i>G</i><sub>H*</sub> ∼ 0 criterion, while they exhibit distinctly different reaction kinetics. Contrary to previous studies that identified surface *O species as active sites, our research reveals that these *O sites are kinetically inert for producing H<sub>2</sub> but are easily reduced to H<sub>2</sub>O. Consequently, the surface Mo atoms, exposed from the rapid reduction of the surface *O species, serve as the actual active sites catalyzing the HER via the Volmer–Heyrovsky mechanism, as confirmed by experimental studies. Our findings highlight the overlooked role of electrostatic repulsion in HER kinetics, a factor not captured by thermodynamic descriptor Δ<i>G</i><sub>H*</sub>. This work provides new insights into the HER mechanism and emphasizes the importance of kinetic investigations for a comprehensive understanding of the HER.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"15 45","pages":"11200–11208 11200–11208"},"PeriodicalIF":4.8000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Overlooked Role of Electrostatic Interactions in HER Kinetics on MXenes: Beyond the Conventional Descriptor ΔG ∼ 0 to Identify the Real Active Site\",\"authors\":\"Xiang Huang, Xiangting Hu, Jiong Wang and Hu Xu*, \",\"doi\":\"10.1021/acs.jpclett.4c0258810.1021/acs.jpclett.4c02588\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Understanding the atomic-level mechanism of the hydrogen evolution reaction (HER) on MXene materials is crucial for developing affordable HER catalysts, while their complex surface terminations present a substantial challenge. Herein, employing constant-potential grand canonical density functional theory calculations, we elucidate the reaction kinetics of the HER on MXenes with various surface terminations by taking experimentally reported Mo<sub>2</sub>C as a prototype. We observe a contradictory scenario on Mo<sub>2</sub>C MXene when using conventional thermodynamic descriptor Δ<i>G</i><sub>H*</sub> (hydrogen binding energy). Both competing surface phases that emerge close to the equilibrium potential meet the Δ<i>G</i><sub>H*</sub> ∼ 0 criterion, while they exhibit distinctly different reaction kinetics. Contrary to previous studies that identified surface *O species as active sites, our research reveals that these *O sites are kinetically inert for producing H<sub>2</sub> but are easily reduced to H<sub>2</sub>O. Consequently, the surface Mo atoms, exposed from the rapid reduction of the surface *O species, serve as the actual active sites catalyzing the HER via the Volmer–Heyrovsky mechanism, as confirmed by experimental studies. Our findings highlight the overlooked role of electrostatic repulsion in HER kinetics, a factor not captured by thermodynamic descriptor Δ<i>G</i><sub>H*</sub>. This work provides new insights into the HER mechanism and emphasizes the importance of kinetic investigations for a comprehensive understanding of the HER.</p>\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"15 45\",\"pages\":\"11200–11208 11200–11208\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpclett.4c02588\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.4c02588","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
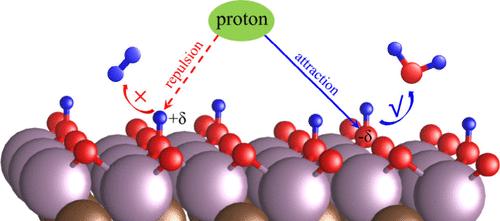
了解 MXene 材料上氢演化反应(HER)的原子级机理对于开发经济实惠的 HER 催化剂至关重要,而其复杂的表面终端则是一个巨大的挑战。在此,我们采用恒电位大规范密度泛函理论计算,以实验报告的 Mo2C 为原型,阐明了具有不同表面端接的 MXene 材料上的氢进化反应动力学。当使用传统的热力学描述符 ΔGH*(氢结合能)时,我们在 Mo2C MXene 上观察到了一种相互矛盾的情况。在接近平衡势时出现的两种竞争表面相都符合 ΔGH* ∼ 0 的标准,但它们的反应动力学却截然不同。与之前将表面 *O 物种确定为活性位点的研究相反,我们的研究揭示了这些 *O 位点在产生 H2 的动力学上是惰性的,但很容易被还原成 H2O。因此,经实验研究证实,因表面 *O 物种快速还原而暴露出来的表面 Mo 原子是通过 Volmer-Heyrovsky 机制催化 HER 的实际活性位点。我们的发现凸显了静电斥力在 HER 动力学中被忽视的作用,而热力学描述符 ΔGH* 并没有捕捉到这一因素。这项工作提供了对 HER 机制的新见解,并强调了动力学研究对全面了解 HER 的重要性。
Overlooked Role of Electrostatic Interactions in HER Kinetics on MXenes: Beyond the Conventional Descriptor ΔG ∼ 0 to Identify the Real Active Site
Understanding the atomic-level mechanism of the hydrogen evolution reaction (HER) on MXene materials is crucial for developing affordable HER catalysts, while their complex surface terminations present a substantial challenge. Herein, employing constant-potential grand canonical density functional theory calculations, we elucidate the reaction kinetics of the HER on MXenes with various surface terminations by taking experimentally reported Mo2C as a prototype. We observe a contradictory scenario on Mo2C MXene when using conventional thermodynamic descriptor ΔGH* (hydrogen binding energy). Both competing surface phases that emerge close to the equilibrium potential meet the ΔGH* ∼ 0 criterion, while they exhibit distinctly different reaction kinetics. Contrary to previous studies that identified surface *O species as active sites, our research reveals that these *O sites are kinetically inert for producing H2 but are easily reduced to H2O. Consequently, the surface Mo atoms, exposed from the rapid reduction of the surface *O species, serve as the actual active sites catalyzing the HER via the Volmer–Heyrovsky mechanism, as confirmed by experimental studies. Our findings highlight the overlooked role of electrostatic repulsion in HER kinetics, a factor not captured by thermodynamic descriptor ΔGH*. This work provides new insights into the HER mechanism and emphasizes the importance of kinetic investigations for a comprehensive understanding of the HER.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


