{"title":"通过高通量第一性原理计算,对电催化硝酸盐还原成氨的高效铜基双原子合金催化剂进行理论预测","authors":"Yuanyuan Wang, Chunmei Tang, Qianlin Li, Ting Xiao and Fujian Xiong","doi":"10.1039/D4TA07470J","DOIUrl":null,"url":null,"abstract":"<p >The electrocatalytic nitrate (NO<small><sub>3</sub></small><small><sup>−</sup></small>) reduction reaction (eNO<small><sub>3</sub></small>RR) to produce valuable ammonia (NH<small><sub>3</sub></small>) is a promising strategy for achieving wastewater treatment and energy conversion. Very recently, dual-atom alloy (DAA) catalysts, featuring active metal dimers embedded in the surface of an inert host metal, have garnered initial research interests owing to the unique geometric and electronic structures of the active metal dimers. The Cu(111) surface shows promising eNO<small><sub>3</sub></small>RR performance, but it still suffers from insufficient activity and selectivity. Herein, stimulated by the concept of DAA, we extensively investigated the potential of Cu-based homonuclear DAAs (TM<small><sub>2</sub></small>Cu, TM = 3d, 4d, 5d) for eNO<small><sub>3</sub></small>RR through high-throughput first-principles calculations. The Mn<small><sub>2</sub></small>Cu, Fe<small><sub>2</sub></small>Cu, and Co<small><sub>2</sub></small>Cu DAAs are found to have excellent eNO<small><sub>3</sub></small>RR performance with lower limiting potentials (<em>U</em><small><sub>L</sub></small>) of −0.18, −0.30, and −0.28 V, respectively, as well as enhanced selectivity, compared with the pristine Cu(111) (<em>U</em><small><sub>L</sub></small> = −0.38 V). The influence of pH and applied potential was further investigated using constant-potential density functional theory (DFT) calculations. Notably, compared to Cu(111), the Mn<small><sub>2</sub></small>Cu and Co<small><sub>2</sub></small>Cu DAAs demonstrate superior activity and selectivity under varying applied potentials and pH conditions. The origin of the activity is attributed to the charge “acceptance–donation” mechanism, which results in continuous d–σ and d–π* interactions between the TM dimer and *NO<small><sub>3</sub></small>, leading to more bonding states near the <em>E</em><small><sub>f</sub></small>, and thus stimulating the adsorption and activation of NO<small><sub>3</sub></small><small><sup>−</sup></small>. The selective enhancement of TM<small><sub>2</sub></small>Cu is due to the fact that NO<small><sub>3</sub></small><small><sup>−</sup></small> gains more electrons from TM dimers compared to H atoms, which helps inhibit the HER. This work not only proposes a new approach for the rational design of efficient eNO<small><sub>3</sub></small>RR catalysts for NH<small><sub>3</sub></small> production by regulating metal active centers, but also provides in-depth insights into the underlying mechanism for the enhanced performance of DAA catalysts.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 5","pages":" 3765-3776"},"PeriodicalIF":9.5000,"publicationDate":"2024-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Theoretical prediction of efficient Cu-based dual-atom alloy catalysts for electrocatalytic nitrate reduction to ammonia via high-throughput first-principles calculations†\",\"authors\":\"Yuanyuan Wang, Chunmei Tang, Qianlin Li, Ting Xiao and Fujian Xiong\",\"doi\":\"10.1039/D4TA07470J\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The electrocatalytic nitrate (NO<small><sub>3</sub></small><small><sup>−</sup></small>) reduction reaction (eNO<small><sub>3</sub></small>RR) to produce valuable ammonia (NH<small><sub>3</sub></small>) is a promising strategy for achieving wastewater treatment and energy conversion. Very recently, dual-atom alloy (DAA) catalysts, featuring active metal dimers embedded in the surface of an inert host metal, have garnered initial research interests owing to the unique geometric and electronic structures of the active metal dimers. The Cu(111) surface shows promising eNO<small><sub>3</sub></small>RR performance, but it still suffers from insufficient activity and selectivity. Herein, stimulated by the concept of DAA, we extensively investigated the potential of Cu-based homonuclear DAAs (TM<small><sub>2</sub></small>Cu, TM = 3d, 4d, 5d) for eNO<small><sub>3</sub></small>RR through high-throughput first-principles calculations. The Mn<small><sub>2</sub></small>Cu, Fe<small><sub>2</sub></small>Cu, and Co<small><sub>2</sub></small>Cu DAAs are found to have excellent eNO<small><sub>3</sub></small>RR performance with lower limiting potentials (<em>U</em><small><sub>L</sub></small>) of −0.18, −0.30, and −0.28 V, respectively, as well as enhanced selectivity, compared with the pristine Cu(111) (<em>U</em><small><sub>L</sub></small> = −0.38 V). The influence of pH and applied potential was further investigated using constant-potential density functional theory (DFT) calculations. Notably, compared to Cu(111), the Mn<small><sub>2</sub></small>Cu and Co<small><sub>2</sub></small>Cu DAAs demonstrate superior activity and selectivity under varying applied potentials and pH conditions. The origin of the activity is attributed to the charge “acceptance–donation” mechanism, which results in continuous d–σ and d–π* interactions between the TM dimer and *NO<small><sub>3</sub></small>, leading to more bonding states near the <em>E</em><small><sub>f</sub></small>, and thus stimulating the adsorption and activation of NO<small><sub>3</sub></small><small><sup>−</sup></small>. The selective enhancement of TM<small><sub>2</sub></small>Cu is due to the fact that NO<small><sub>3</sub></small><small><sup>−</sup></small> gains more electrons from TM dimers compared to H atoms, which helps inhibit the HER. This work not only proposes a new approach for the rational design of efficient eNO<small><sub>3</sub></small>RR catalysts for NH<small><sub>3</sub></small> production by regulating metal active centers, but also provides in-depth insights into the underlying mechanism for the enhanced performance of DAA catalysts.</p>\",\"PeriodicalId\":82,\"journal\":{\"name\":\"Journal of Materials Chemistry A\",\"volume\":\" 5\",\"pages\":\" 3765-3776\"},\"PeriodicalIF\":9.5000,\"publicationDate\":\"2024-11-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Materials Chemistry A\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d4ta07470j\",\"RegionNum\":2,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d4ta07470j","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Theoretical prediction of efficient Cu-based dual-atom alloy catalysts for electrocatalytic nitrate reduction to ammonia via high-throughput first-principles calculations†
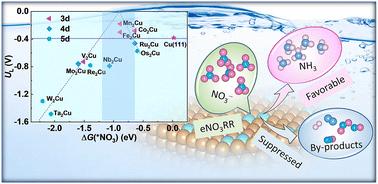
The electrocatalytic nitrate (NO3−) reduction reaction (eNO3RR) to produce valuable ammonia (NH3) is a promising strategy for achieving wastewater treatment and energy conversion. Very recently, dual-atom alloy (DAA) catalysts, featuring active metal dimers embedded in the surface of an inert host metal, have garnered initial research interests owing to the unique geometric and electronic structures of the active metal dimers. The Cu(111) surface shows promising eNO3RR performance, but it still suffers from insufficient activity and selectivity. Herein, stimulated by the concept of DAA, we extensively investigated the potential of Cu-based homonuclear DAAs (TM2Cu, TM = 3d, 4d, 5d) for eNO3RR through high-throughput first-principles calculations. The Mn2Cu, Fe2Cu, and Co2Cu DAAs are found to have excellent eNO3RR performance with lower limiting potentials (UL) of −0.18, −0.30, and −0.28 V, respectively, as well as enhanced selectivity, compared with the pristine Cu(111) (UL = −0.38 V). The influence of pH and applied potential was further investigated using constant-potential density functional theory (DFT) calculations. Notably, compared to Cu(111), the Mn2Cu and Co2Cu DAAs demonstrate superior activity and selectivity under varying applied potentials and pH conditions. The origin of the activity is attributed to the charge “acceptance–donation” mechanism, which results in continuous d–σ and d–π* interactions between the TM dimer and *NO3, leading to more bonding states near the Ef, and thus stimulating the adsorption and activation of NO3−. The selective enhancement of TM2Cu is due to the fact that NO3− gains more electrons from TM dimers compared to H atoms, which helps inhibit the HER. This work not only proposes a new approach for the rational design of efficient eNO3RR catalysts for NH3 production by regulating metal active centers, but also provides in-depth insights into the underlying mechanism for the enhanced performance of DAA catalysts.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


