{"title":"评估用于枯木分解微生物群落分析的 DNA 提取方法。","authors":"Yanmei Zhang, Zewei Song, Jonathan S. Schilling","doi":"10.1002/mbo3.70007","DOIUrl":null,"url":null,"abstract":"<p>As technologies advance alongside metabarcoding and metagenomic resources, particularly for larger fungal genomes, DNA extraction methods must be optimized to meet higher thresholds, especially from complex environmental substrates. This study focused on extracting fungal genomic compounds from woody substrates, a challenge due to the embedment of endophytic and saprotrophic fungi within wood cells, the physical recalcitrance of wood, the adsorption of nucleic acids to wood polymers, and the release of downstream inhibitors. Hypothesizing that cetyltrimethylammonium bromide would be the best option, we compared prominent methods by extracting and sequencing microbial DNA from sound and decayed birch (<i>Betula papyrifera</i>) and pine (<i>Pinus resinosa</i>). DNA quantities varied significantly depending on extraction methods and decay stage. The quality of DNA, in terms of purity and integrity, significantly impacted whether the samples could be amplified and sequenced. However, amplicon sequencing of bacterial and fungal communities revealed no significant extraction bias. This, along with the sequencing effectiveness and cost/time efficiency, indicates that Qiagen is the gold standard for woody substrates. This study increases confidence in published amplicon data sets regardless of the extraction methods, provides a cost-benefit table for making protocol decisions, and offers guidance on fungal DNA extractions from complex organic substrates (sound and decayed wood) that would best suit future metagenomic efforts.</p>","PeriodicalId":18573,"journal":{"name":"MicrobiologyOpen","volume":"13 6","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2024-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11561137/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evaluation of DNA Extraction Methods for Microbial Community Profiling in Deadwood Decomposition\",\"authors\":\"Yanmei Zhang, Zewei Song, Jonathan S. Schilling\",\"doi\":\"10.1002/mbo3.70007\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>As technologies advance alongside metabarcoding and metagenomic resources, particularly for larger fungal genomes, DNA extraction methods must be optimized to meet higher thresholds, especially from complex environmental substrates. This study focused on extracting fungal genomic compounds from woody substrates, a challenge due to the embedment of endophytic and saprotrophic fungi within wood cells, the physical recalcitrance of wood, the adsorption of nucleic acids to wood polymers, and the release of downstream inhibitors. Hypothesizing that cetyltrimethylammonium bromide would be the best option, we compared prominent methods by extracting and sequencing microbial DNA from sound and decayed birch (<i>Betula papyrifera</i>) and pine (<i>Pinus resinosa</i>). DNA quantities varied significantly depending on extraction methods and decay stage. The quality of DNA, in terms of purity and integrity, significantly impacted whether the samples could be amplified and sequenced. However, amplicon sequencing of bacterial and fungal communities revealed no significant extraction bias. This, along with the sequencing effectiveness and cost/time efficiency, indicates that Qiagen is the gold standard for woody substrates. This study increases confidence in published amplicon data sets regardless of the extraction methods, provides a cost-benefit table for making protocol decisions, and offers guidance on fungal DNA extractions from complex organic substrates (sound and decayed wood) that would best suit future metagenomic efforts.</p>\",\"PeriodicalId\":18573,\"journal\":{\"name\":\"MicrobiologyOpen\",\"volume\":\"13 6\",\"pages\":\"\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2024-11-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11561137/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"MicrobiologyOpen\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/mbo3.70007\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"MicrobiologyOpen","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mbo3.70007","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
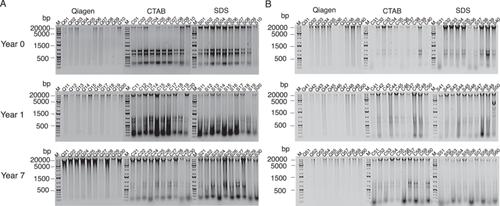
随着新陈代谢编码和元基因组资源技术的发展,尤其是对于较大的真菌基因组而言,DNA 提取方法必须进行优化,以满足更高的要求,尤其是从复杂的环境基质中提取。这项研究的重点是从木质基质中提取真菌基因组化合物,这是一项挑战,因为内生真菌和嗜渍真菌会嵌入木质细胞、木质的物理不稳定性、核酸对木质聚合物的吸附以及下游抑制剂的释放。我们推测十六烷基三甲基溴化铵将是最佳选择,并通过从完好和腐烂的桦树(Betula papyrifera)和松树(Pinus resinosa)中提取微生物 DNA 并对其进行测序,对主要方法进行了比较。DNA数量因提取方法和腐烂阶段的不同而有很大差异。DNA 的纯度和完整性对样本能否扩增和测序有很大影响。不过,细菌和真菌群落的扩增子测序结果显示,提取方法没有明显偏差。这一点以及测序效果和成本/时间效率表明,对于木质基质来说,Qiagen 是黄金标准。这项研究提高了人们对已发表的扩增子数据集的信心,无论其提取方法如何,并为制定方案提供了成本效益表,为从复杂的有机基质(健全和腐朽的木材)中提取真菌 DNA 提供了指导,最适合未来的元基因组研究工作。
Evaluation of DNA Extraction Methods for Microbial Community Profiling in Deadwood Decomposition
As technologies advance alongside metabarcoding and metagenomic resources, particularly for larger fungal genomes, DNA extraction methods must be optimized to meet higher thresholds, especially from complex environmental substrates. This study focused on extracting fungal genomic compounds from woody substrates, a challenge due to the embedment of endophytic and saprotrophic fungi within wood cells, the physical recalcitrance of wood, the adsorption of nucleic acids to wood polymers, and the release of downstream inhibitors. Hypothesizing that cetyltrimethylammonium bromide would be the best option, we compared prominent methods by extracting and sequencing microbial DNA from sound and decayed birch (Betula papyrifera) and pine (Pinus resinosa). DNA quantities varied significantly depending on extraction methods and decay stage. The quality of DNA, in terms of purity and integrity, significantly impacted whether the samples could be amplified and sequenced. However, amplicon sequencing of bacterial and fungal communities revealed no significant extraction bias. This, along with the sequencing effectiveness and cost/time efficiency, indicates that Qiagen is the gold standard for woody substrates. This study increases confidence in published amplicon data sets regardless of the extraction methods, provides a cost-benefit table for making protocol decisions, and offers guidance on fungal DNA extractions from complex organic substrates (sound and decayed wood) that would best suit future metagenomic efforts.
期刊介绍:
MicrobiologyOpen is a peer reviewed, fully open access, broad-scope, and interdisciplinary journal delivering rapid decisions and fast publication of microbial science, a field which is undergoing a profound and exciting evolution in this post-genomic era.
The journal aims to serve the research community by providing a vehicle for authors wishing to publish quality research in both fundamental and applied microbiology. Our goal is to publish articles that stimulate discussion and debate, as well as add to our knowledge base and further the understanding of microbial interactions and microbial processes.
MicrobiologyOpen gives prompt and equal consideration to articles reporting theoretical, experimental, applied, and descriptive work in all aspects of bacteriology, virology, mycology and protistology, including, but not limited to:
- agriculture
- antimicrobial resistance
- astrobiology
- biochemistry
- biotechnology
- cell and molecular biology
- clinical microbiology
- computational, systems, and synthetic microbiology
- environmental science
- evolutionary biology, ecology, and systematics
- food science and technology
- genetics and genomics
- geobiology and earth science
- host-microbe interactions
- infectious diseases
- natural products discovery
- pharmaceutical and medicinal chemistry
- physiology
- plant pathology
- veterinary microbiology
We will consider submissions across unicellular and cell-cluster organisms: prokaryotes (bacteria, archaea) and eukaryotes (fungi, protists, microalgae, lichens), as well as viruses and prions infecting or interacting with microorganisms, plants and animals, including genetic, biochemical, biophysical, bioinformatic and structural analyses.
The journal features Original Articles (including full Research articles, Method articles, and Short Communications), Commentaries, Reviews, and Editorials. Original papers must report well-conducted research with conclusions supported by the data presented in the article. We also support confirmatory research and aim to work with authors to meet reviewer expectations.
MicrobiologyOpen publishes articles submitted directly to the journal and those referred from other Wiley journals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


