{"title":"适用于分布式多 GPU 架构的高效 RI-MP2 算法","authors":"Calum Snowdon, and , Giuseppe M. J. Barca*, ","doi":"10.1021/acs.jctc.4c0081410.1021/acs.jctc.4c00814","DOIUrl":null,"url":null,"abstract":"<p >Second-order Møller–Plesset perturbation theory (MP2) using the Resolution of the Identity approximation (RI-MP2) is a widely used method for computing molecular energies beyond the Hartree–Fock mean-field approximation. However, its high computational cost and lack of efficient algorithms for modern supercomputing architectures limit its applicability to large molecules. In this paper, we present the first distributed-memory many-GPU RI-MP2 algorithm explicitly designed to utilize hundreds of GPU accelerators for every step of the computation. Our novel algorithm achieves near-peak performance on GPU-based supercomputers through the development of a distributed memory algorithm for forming RI-MP2 intermediate tensors with zero internode communication, except for a single <i></i><math><mi>O</mi><mrow><mo>(</mo><msup><mi>N</mi><mn>2</mn></msup><mo>)</mo></mrow></math> asynchronous broadcast, and a distributed memory algorithm for the <i></i><math><mi>O</mi><mrow><mo>(</mo><msup><mi>N</mi><mn>5</mn></msup><mo>)</mo></mrow></math> energy reduction step, capable of sustaining near-peak performance on clusters with several hundred GPUs. Comparative analysis shows our implementation outperforms state-of-the-art quantum chemistry software by over 3.5 times in speed while achieving an 8-fold reduction in computational power consumption. Benchmarking on the Perlmutter supercomputer, our algorithm achieves 11.8 PFLOP/s (83% of peak performance) performing and the RI-MP2 energy calculation on a 314-water cluster with 7850 primary and 30,144 auxiliary basis functions in 4 min on 180 nodes and 720 A100 GPUs. This performance represents a substantial improvement over traditional CPU-based methods, demonstrating significant time-to-solution and power consumption benefits of leveraging modern GPU-accelerated computing environments for quantum chemistry calculations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 21","pages":"9394–9406 9394–9406"},"PeriodicalIF":5.7000,"publicationDate":"2024-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"An Efficient RI-MP2 Algorithm for Distributed Many-GPU Architectures\",\"authors\":\"Calum Snowdon, and , Giuseppe M. J. Barca*, \",\"doi\":\"10.1021/acs.jctc.4c0081410.1021/acs.jctc.4c00814\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Second-order Møller–Plesset perturbation theory (MP2) using the Resolution of the Identity approximation (RI-MP2) is a widely used method for computing molecular energies beyond the Hartree–Fock mean-field approximation. However, its high computational cost and lack of efficient algorithms for modern supercomputing architectures limit its applicability to large molecules. In this paper, we present the first distributed-memory many-GPU RI-MP2 algorithm explicitly designed to utilize hundreds of GPU accelerators for every step of the computation. Our novel algorithm achieves near-peak performance on GPU-based supercomputers through the development of a distributed memory algorithm for forming RI-MP2 intermediate tensors with zero internode communication, except for a single <i></i><math><mi>O</mi><mrow><mo>(</mo><msup><mi>N</mi><mn>2</mn></msup><mo>)</mo></mrow></math> asynchronous broadcast, and a distributed memory algorithm for the <i></i><math><mi>O</mi><mrow><mo>(</mo><msup><mi>N</mi><mn>5</mn></msup><mo>)</mo></mrow></math> energy reduction step, capable of sustaining near-peak performance on clusters with several hundred GPUs. Comparative analysis shows our implementation outperforms state-of-the-art quantum chemistry software by over 3.5 times in speed while achieving an 8-fold reduction in computational power consumption. Benchmarking on the Perlmutter supercomputer, our algorithm achieves 11.8 PFLOP/s (83% of peak performance) performing and the RI-MP2 energy calculation on a 314-water cluster with 7850 primary and 30,144 auxiliary basis functions in 4 min on 180 nodes and 720 A100 GPUs. This performance represents a substantial improvement over traditional CPU-based methods, demonstrating significant time-to-solution and power consumption benefits of leveraging modern GPU-accelerated computing environments for quantum chemistry calculations.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 21\",\"pages\":\"9394–9406 9394–9406\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-10-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00814\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00814","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
An Efficient RI-MP2 Algorithm for Distributed Many-GPU Architectures
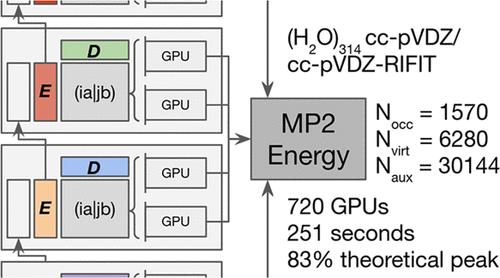
Second-order Møller–Plesset perturbation theory (MP2) using the Resolution of the Identity approximation (RI-MP2) is a widely used method for computing molecular energies beyond the Hartree–Fock mean-field approximation. However, its high computational cost and lack of efficient algorithms for modern supercomputing architectures limit its applicability to large molecules. In this paper, we present the first distributed-memory many-GPU RI-MP2 algorithm explicitly designed to utilize hundreds of GPU accelerators for every step of the computation. Our novel algorithm achieves near-peak performance on GPU-based supercomputers through the development of a distributed memory algorithm for forming RI-MP2 intermediate tensors with zero internode communication, except for a single asynchronous broadcast, and a distributed memory algorithm for the energy reduction step, capable of sustaining near-peak performance on clusters with several hundred GPUs. Comparative analysis shows our implementation outperforms state-of-the-art quantum chemistry software by over 3.5 times in speed while achieving an 8-fold reduction in computational power consumption. Benchmarking on the Perlmutter supercomputer, our algorithm achieves 11.8 PFLOP/s (83% of peak performance) performing and the RI-MP2 energy calculation on a 314-water cluster with 7850 primary and 30,144 auxiliary basis functions in 4 min on 180 nodes and 720 A100 GPUs. This performance represents a substantial improvement over traditional CPU-based methods, demonstrating significant time-to-solution and power consumption benefits of leveraging modern GPU-accelerated computing environments for quantum chemistry calculations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


