通过 Ab Initio 大卡农蒙特卡洛揭示 RuP2 上的电催化氢气进化反应途径
IF 13.1
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
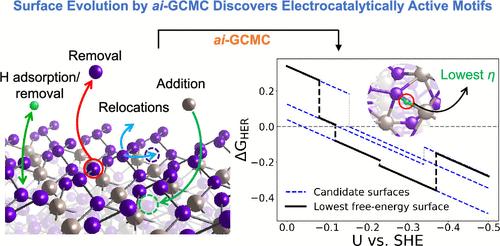
在本研究中,通过对电催化氢进化反应(HER)的第一原理密度泛函理论(DFT)计算,确定了磷化钌(RuP2)的高催化反应活性。通过应用 ab initio grand canonical Monte Carlo (ai-GCMC) 算法考虑了复杂的表面重构,从而有效地对可能的表面进行了充分的相空间探索。结合表相普尔贝图,我们能够确定在特定实验环境下获得的实际表面,从而更准确地了解活性位点的性质和吸附剂的结合强度。具体来说,通过使用 ai-GCMC 生成的数百个表面重构和氢化状态,我们确定了 RuP2 在酸性水溶液条件下最有利的表面相。我们发现,在一个狭窄的电极电位窗口内,氢反应活性由具有不同化学计量学的多个表面决定。在酸性条件下,我们发现了高 H 覆盖率的重构表面,由附加 Ru 原子引入或由 P 空位暴露的表面 Ru 位点因其几乎可逆的 H 结合而成为 HER 的活性位点。这项研究通过探索电催化剂的动态表面相,从原子学角度揭示了 RuP2 上高 HER 活性的起源,并提出了一种可通用的方法,用于探索重构/氢化表面空间与实验条件的函数关系。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Unveiling the Electrocatalytic Hydrogen Evolution Reaction Pathway on RuP2 through Ab Initio Grand Canonical Monte Carlo
In this study, the high catalytic reactivity of ruthenium phosphide (RuP2) has been identified by first-principles density functional theory (DFT) calculations for the electrocatalytic hydrogen evolution reaction (HER). Complex surface reconstructions are considered by applying the ab initio grand canonical Monte Carlo (ai-GCMC) algorithm, efficiently providing a sufficient phase-space exploration of possible surfaces. Combined with surface-phase Pourbaix diagrams, we are able to identify the actual surfaces that obtained under specific experimental environments, thus leading to a more accurate understanding of the nature of the active sites and the binding strength of adsorbates. Specifically, through hundreds of surface reconstructions and hydrogenation states generated with ai-GCMC, we identify the most favorable surface phases of RuP2 under aqueous acidic conditions. We discover that the HER activity is determined by multiple surfaces with different stoichiometries within a narrow electrode potential window. Low HER overpotential (η) has been found for each of the identified surfaces, as low as 0.04 V. High H-coverage reconstructed surfaces have been discovered under acidic conditions, and the surface Ru sites introduced by additional Ru adatoms or exposed by P-vacancies serve as the active sites for HER based on their nearly reversible H binding. This work provides atomistic insights into the origin of high HER activity on RuP2 by exploring the dynamic surface phases of electrocatalysts and features a generalizable method to explore the reconstructed/hydrogenated surface space as a function of experimental conditions.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


