以 FBPase 的 AMP 位点为目标的亲和力/共价键双驱动抑制剂的结构引导设计
IF 6.8
1区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
果糖-1,6-二磷酸酶(FBPase)作为一种与癌症和 II 型糖尿病相关的靶点引起了人们的极大兴趣。以 AMP 异构位点为靶点的 FBPase 抑制剂已有文献记载,但其有限的选择性引起了人们对不良反应的担忧。为了解决这个问题,我们根据对 FBPase 的 AMP 口袋和邻近半胱氨酸残基(C179)的药效学知识,利用半胱氨酸靶向反应性弹头筛选,然后采用结构优化策略,设计出了亲和力/共价键双驱动抑制剂。牵引和 Western 印迹检测证实 FBPase 是肝细胞的直接靶标。FBPase-11 的 X 射线共晶体结构和 Cov_DOX 计算表明,氢键和 π-π 堆积是磺酰脲类 FBPase 共价抑制剂抑制作用的主要驱动力,而与 C179 的共价结合则增强了抑制剂的持久降糖效果。总之,这项工作凸显了亲和力/共价键双驱动抑制剂在药物开发中的潜力,并为开发针对 AMP 相关蛋白的强效药物提供了一种前景广阔的方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
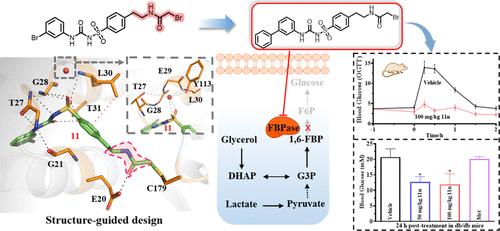
Structure-Guided Design of Affinity/Covalent-Bond Dual-Driven Inhibitors Targeting the AMP Site of FBPase
Fructose-1,6-bisphosphatase (FBPase) has attracted substantial interest as a target associated with cancer and type II diabetes. FBPase inhibitors targeting the AMP allosteric site have been documented, but their limited selectivity has raised concerns about adverse effects. To address this issue, we designed the affinity/covalent-bond dual-driven inhibitors based on the pharmacophore knowledge of the AMP pocket and neighboring cysteine residue (C179) of FBPase using the cysteine-targeting reactivity warhead screen followed by a structural optimization strategy. Pull-down and Western Blotting assays confirmed FBPase as a direct target in hepatic cells. X-ray cocrystallographic structure of FBPase-11 and Cov_DOX calculation demonstrated that hydrogen bonding and π–π stacking were the predominant driving force for the inhibition of sulfonylurea-based FBPase covalent inhibitors, while covalent binding with C179 enhances the inhibitors’ long-lasting hypoglycemic effects. Together, this work highlights the potential of affinity/covalent-bond dual-driven inhibitors in drug development and provides a promising approach for developing potent drugs targeting AMP-associated proteins.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Medicinal Chemistry
医学-医药化学
CiteScore
4.00
自引率
11.00%
发文量
804
审稿时长
1.9 months
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


