多组分合成有助于发现作为组蛋白去乙酰化酶抑制剂的新型奎司他衍生化学类型
IF 5.9
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
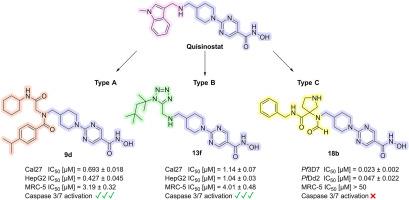
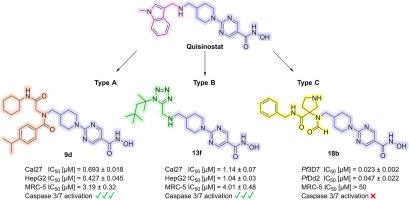
在这项研究中,我们合成并评估了源自临床候选药物喹司他的新型组蛋白去乙酰化酶(HDAC)抑制剂。我们利用多组分反应(MCR)快速生成了一个由 16 种化合物组成的化合物库,这些化合物分为三种新的化学类型,从而实现了高效的结构-活性关系研究。首先,评估了这些化合物对恶性疟原虫菌株 3D7 和 Dd2(主要的疟疾致病寄生虫)的活性,确定 C 型系列的 18b 化合物最有效。它显示出较低的纳摩尔 IC50 值(IC50 (3D7) = 0.023 μM;IC50 (Dd2) = 0.047 μM)和较高的寄生虫选择性(SIMRC-5/Pf3D7 > 2174)。HDAC 抑制试验证实,它对恶性疟原虫酶 PfHDAC1(IC50 = 0.037 μM)以及人类 HDAC1(IC50 = 0.021 μM)和 HDAC6(IC50 = 0.25 μM)有显著抑制作用。对接研究表明,18b 与恶性疟原虫和人类 HDAC1 的结合模式不同。此外,还在 Cal27(头颈癌)、HepG2(肝癌)、A2780(卵巢癌)和 U87(胶质母细胞瘤)细胞系中评估了其体外抗癌活性。与 18b 相反,化合物 9b、9d 和 13f 显示出强大的抗增殖活性和 caspase 3/7 激活作用。此外,这些化合物还能引起组蛋白 H3 和 α-微管蛋白的超乙酰化,表明它们具有强大的细胞靶标参与能力。总之,在这项工作中,我们发现了具有选择性抗疟活性的 HDAC 抑制剂 18b,以及具有选择性抗癌活性的 9b、9d 和 13f,为进一步的药物开发工作提供了有价值的线索,目的是创造出与喹司他相比对非癌细胞细胞毒性更低的衍生物。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Multicomponent syntheses enable the discovery of novel quisinostat-derived chemotypes as histone deacetylase inhibitors
In this study, we synthesized and evaluated novel histone deacetylase (HDAC) inhibitors derived from the clinical candidate quisinostat. A library of 16 compounds categorized in three novel chemotypes was rapidly generated using multicomponent reactions (MCRs), enabling efficient structure-activity relationship studies. First, the compounds were evaluated for their activity against the Plasmodium falciparum strains 3D7 and Dd2, the main malaria-causing parasite, identifying compound 18b of the type C series as the most potent. It demonstrated low nanomolar IC50 values (IC50 (3D7) = 0.023 μM; IC50 (Dd2) = 0.047 μM) and high parasite selectivity (SIMRC−5/Pf3D7 > 2174). HDAC inhibition assays confirmed substantial inhibition of the P. falciparum enzyme PfHDAC1 (IC50 = 0.037 μM) as well as of human HDAC1 (IC50 = 0.021 μM) and HDAC6 (IC50 = 0.25 μM). Docking studies suggested distinct binding modes of 18b in P. falciparum and human HDAC1. Additionally, the in vitro anticancer activity was evaluated in Cal27 (head-neck carcinoma), HepG2 (hepatocellular carcinoma), A2780 (ovarian carcinoma), and U87 (glioblastoma) cell lines. Compounds 9b, 9d, and 13f showed potent antiproliferative activity and caspase 3/7 activation, in contrast to 18b. Furthermore, these compounds caused hyperacetylation of histone H3 and α-tubulin, indicating robust cellular target engagement. Overall, in this work we have identified the HDAC inhibitor 18b with selective antiplasmodial and 9b, 9d, and 13f with selective anticancer activities, providing valuable hits for further drug development efforts aimed at creating derivatives with reduced cytotoxicity against non-cancer cells compared to quisinostat.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


