高爆炸晶体密度和性能的机器学习模型
IF 7.2
2区 材料科学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
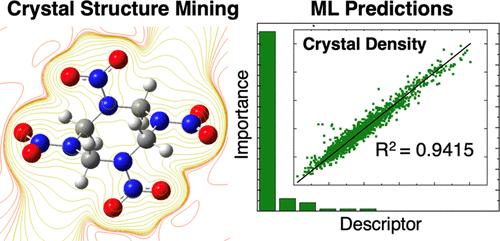
具有高能量密度和高性能的新型炸药的发现速度基本停滞不前。通过机器学习进行快速特性预测,有可能在合成新分子之前对大量分子进行筛选,从而加速新分子的发现。为了实现这一目标,我们建立了一个包含 21,000 个分子的数据库,其中包含高能官能团的实验合成分子。利用实验密度测量和高通量电子结构与原子计算相结合的方法,我们计算出了所有 21,000 种化合物的引爆速度和压力。利用这些数值,我们训练了预测密度、爆速和爆压的机器学习模型。值得注意的是,我们的晶体密度模型超过了目前所有模型的准确性,并将之前最佳模型的均方根误差 (RMSE) 降低了 20%。与过去的研究相比,我们的模型性能有了很大提高,这要归功于我们处理了手性指定的简化分子输入线输入系统(SMILES)字符串,并引入了新的分子描述符 MolDensity。为了阐明描述符的重要性,我们评估了可解释描述符的重要性,并将统计驱动的机器学习模型的准确性与通常假定控制材料密度的描述符组成的模型进行了比较。我们的模型成本低廉,但预测准确性很高,因此未来可以创建人工智能(AI)模型,对大量(106 种)化合物进行筛选,找出在晶体密度、爆速和爆压方面性能最高的化合物。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Machine Learning Models for High Explosive Crystal Density and Performance
The rate of discovery of new explosives with superior energy density and performance has largely stalled. Rapid property prediction through machine learning has the potential to accelerate the discovery of new molecules by screening of large numbers of molecules before they are ever synthesized. To support this goal, we assembled a 21,000-molecule database of experimentally synthesized molecules containing energetic functional groups. Using a combination of experimental density measurements and high throughput electronic structure and atomistic calculations, we calculated detonation velocities and pressures for all 21,000 compounds. Using these values, we trained machine learning models for the prediction of density, detonation velocity and detonation pressure. Notably, our model for crystal density surpassed the accuracy of all current models and decreased the root-mean square error (RMSE) of the previous best model by 20%. This improvement in model performance relative to past works is attributed to our handling of chiral-specified Simplified Molecular-Input Line-Entry System (SMILES) strings and introduction of a new molecular descriptor, MolDensity. To elucidate descriptor importance, we evaluated interpretable descriptors in terms of importance and compared the accuracy of a statistics-driven machine learning model against a model comprised of descriptors typically assumed to control material density. The inexpensive, yet highly accurate predictions from our models should enable creation of future artificial intelligence (AI) models that are able to screen large numbers (>106) of compounds to find the highest performing compounds in terms of crystal density, detonation velocity and detonation pressure.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Chemistry of Materials
工程技术-材料科学:综合
CiteScore
14.10
自引率
5.80%
发文量
929
审稿时长
1.5 months
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


