基于 MXene 的单原子催化剂的价电子匹配定律
IF 13.1
1区 化学
Q1 Energy
引用次数: 0
摘要
单原子催化剂(SAC)因其可调节的催化活性而在能源和环境科学领域引起了广泛关注。在本研究中,我们研究了 MXene-anchored SACs(M-Ti2C/M-Ti2CO2)中单原子与吸附中间产物(O、N、C 和 H)之间价电子数的匹配。密度泛函理论结果表明,M-Ti2C 中界面掺杂金属的价电子数(VM)与吸附中间产物的价电子数(VA)之和遵循 10 价电子匹配定律。此外,根据 10 价电子匹配定律,我们推断出 M-Ti2CO2 中分子吸附中间体相互作用的价电子数(k)和 VM 之和遵循 11 价电子匹配定律。M-Ti2CO2 和 H2O 中界面电子之间的静电排斥削弱了中间产物的吸附。此外,我们还应用 11 价电子匹配定律来指导氮还原反应催化剂的设计,特别是 M-Ti2CO2 结构中 N2 → NNH 转化催化剂的设计。我们使用确定的独立性筛选和稀疏化算子算法拟合了一个简单的吸附剂三维描述符(R2 高达 0.970),用于催化剂设计。我们的研究为基于 MXene 的催化剂引入了掺杂金属(单原子)和吸附中间体(原子和分子)之间的价电子匹配原理,为高性能 SAC 的设计提供了新的见解。本文章由计算机程序翻译,如有差异,请以英文原文为准。
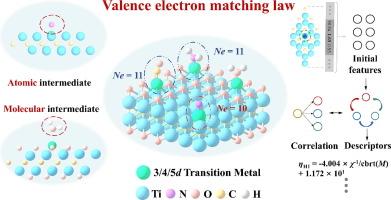
Valence electron matching law for MXene-based single-atom catalysts
Single-atom catalysts (SACs) have attracted considerable interest in the fields of energy and environmental science due to their adjustable catalytic activity. In this study, we investigated the matching of valence electron numbers between single atoms and adsorbed intermediates (O, N, C, and H) in MXene-anchored SACs (M-Ti2C/M-Ti2CO2). The density functional theory results demonstrated that the sum of the valence electron number (VM) of the interface-doped metal and the valence electron number (VA) of the adsorbed intermediates in M-Ti2C followed the 10-valence electron matching law. Furthermore, based on the 10-valence electron matching law, we deduced that the sum of the valence electron number (k) and VM for the molecular adsorption intermediate interactions in M-Ti2CO2 adhered to the 11-valence electron matching law. Electrostatic repulsion between the interface electrons in M-Ti2CO2 and H2O weakened the adsorption of intermediates. Furthermore, we applied the 11-valence electron matching law to guide the design of catalysts for nitrogen reduction reaction, specifically for N2 → NNH conversion, in the M-Ti2CO2 structure. The sure independence screening and sparsifying operator algorithm was used to fit a simple three-dimensional descriptor of the adsorbate (R2 up to 0.970) for catalyst design. Our study introduced a valence electron matching principle between doped metals (single atoms) and adsorbed intermediates (atomic and molecular) for MXene-based catalysts, providing new insights into the design of high-performance SACs.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Energy Chemistry
CHEMISTRY, APPLIED-CHEMISTRY, PHYSICAL
CiteScore
19.10
自引率
8.40%
发文量
3631
审稿时长
15 days
期刊介绍:
The Journal of Energy Chemistry, the official publication of Science Press and the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, serves as a platform for reporting creative research and innovative applications in energy chemistry. It mainly reports on creative researches and innovative applications of chemical conversions of fossil energy, carbon dioxide, electrochemical energy and hydrogen energy, as well as the conversions of biomass and solar energy related with chemical issues to promote academic exchanges in the field of energy chemistry and to accelerate the exploration, research and development of energy science and technologies.
This journal focuses on original research papers covering various topics within energy chemistry worldwide, including:
Optimized utilization of fossil energy
Hydrogen energy
Conversion and storage of electrochemical energy
Capture, storage, and chemical conversion of carbon dioxide
Materials and nanotechnologies for energy conversion and storage
Chemistry in biomass conversion
Chemistry in the utilization of solar energy
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


