基于门的量子计算机中的高自由度蛋白质结构预测
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
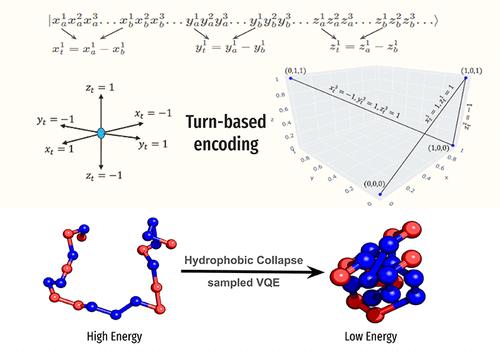
蛋白质折叠是根据氨基酸序列追溯蛋白质三维(3D)结构的过程,是生物学中一个具有半个世纪历史的难题。蛋白质的功能与其结构息息相关,因此有必要研究蛋白质折叠,以便更好地了解细胞和分子过程。虽然最近基于人工智能的方法在蛋白质结构预测方面取得了巨大成功,但当蛋白质的序列相似度较低时,其准确性就会降低。经典模拟在生成广泛的构象采样方面面临挑战。在这项工作中,我们开发了一种具有更多自由度的新型回合编码算法,它能成功地在基于门的量子计算机上运行,并利用多达 114 量子位(IBM 硬件)预测不同长度的蛋白质结构。为了使量子计算机能够解决这个问题,蛋白质序列采用了简单的 HP 模型(H = 疏水残基,P = 极性残基)进行描述。所提出的公式成功地捕捉到了蛋白质折叠中所谓的成核步骤--疏水塌缩,它将疏水残基带到了蛋白质的核心。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Protein Structure Prediction with High Degrees of Freedom in a Gate-Based Quantum Computer
Protein folding, which traces the protein three-dimensional (3D) structure from its amino acid sequence, is a half-a-century-old problem in biology. The function of the protein correlates with its structure, emphasizing the need to study protein folding to understand the cellular and molecular processes better. While recent AI-based methods have shown significant success in protein structure prediction, their accuracy diminishes with proteins of low sequence similarity. Classical simulations face challenges in generating extensive conformational samplings. In this work, we develop a novel turn-based encoding algorithm with more significant degrees of freedom that successfully runs on a gate-based quantum computer and predicts the structure of proteins of varied lengths utilizing up to 114 qubits (IBM hardware). To make the problem tractable in quantum computers, the protein sequences were described with the simplistic HP model (H = hydrophobic residues, P = polar residues). The proposed formulation successfully captures the so-called nucleation step in protein folding, the hydrophobic collapse, that brings the hydrophobic residues to the core of the protein.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


