价电子和配位结构引导下的硫化碳双键水解裂解金属活性位点设计
IF 11.3
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
在金属位点上催化裂解碳硫(C═S)双键而不发生失活现象引起了基础催化研究和工业化学的极大兴趣。本文通过机器学习和密度泛函理论(DFT)计算开发了活性描述符,用于筛选过渡金属单位催化剂,量化原子电子特性和配位构型对碳硫双键水解的影响。研究发现,活性位点的价电子数和电负性与 C═S 活化和硫中毒密切相关,其中铁在一系列金属中心中表现出较高的催化潜力。另一方面,孤立的 Fe1 和 Fe2 位点有利于硫化羰基(COS)的吸附和活化,而 COS 在 Fe3 中空位点上很容易解离成 *S 和 *CO,从而形成牢固的 Fe-S 键,导致催化剂失活。正如预期的那样,按设计的 Fe1-N4 位点在 100 °C 时实现了约 96% 的 COS 转化率,略高于 Fe2-N4 位点,约为 Fe/C 位点的 8 倍,也优于其他单原子催化剂(如 Co-NC、Ni-NC、Sn-NC 和 Bi-NC)。原位表征和理论计算相结合表明,*COS 和 *H2O/*OH 在 Fe-N4 位点上具有竞争吸附关系,两个 Fe-N4 位点可以通过溢出的 H 和 OH 协同催化 COS 的水解。本文章由计算机程序翻译,如有差异,请以英文原文为准。
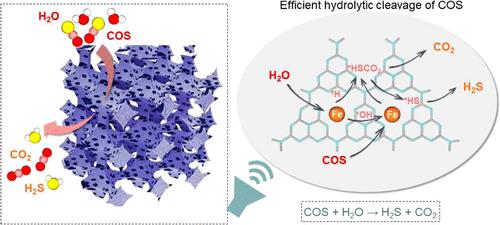
Valence Electron and Coordination Structure Guided Metal Active Site Design for Hydrolytic Cleavage of Carbon–Sulfide Double Bonds
The catalytic cleavage of carbon–sulfur (C═S) double bonds on the metal sites without deactivation has aroused great interest in both fundamental catalytic research and industrial chemistry. Herein, activity descriptors are developed via machine learning and density functional theory (DFT) calculations to screen transition-metal single-site catalysts, which quantify the effect of both atomic electronic properties and coordination configuration on the hydrolysis of C═S double bonds. The valence electron number and electronegativity of active sites are found to be well related to C═S activation and sulfur poisoning, where Fe demonstrates high catalytic potential among a series of metal centers. On the other hand, the isolated Fe1 and Fe2 sites favor carbonyl sulfide (COS) adsorption and activation, while the COS easily dissociates into *S and *CO on Fe3 hollow site, thus resulting in the formation of robust Fe–S bonds and catalyst deactivation. As anticipated, the as-designed Fe1–N4 site achieves a COS conversion of ca. 96% at 100 °C, slightly better than the Fe2–N4 site, approximately 8 times higher than that of the Fe/C, which is also better than those of other monatomic catalysts (such as Co-NC, Ni-NC, Sn-NC, and Bi-NC). The combination of in situ characterizations and theoretical calculations suggests that *COS and *H2O/*OH have a competitive adsorption relationship on Fe–N4 sites, and two Fe–N4 sites can synergistically catalyze the COS hydrolysis through the spilled H and OH.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


