Wei Wang, Jun Min, Qinghua Luo, Xunhu Gu, Min Li, Xu Liu
{"title":"赖氨酸乙酰转移酶 TIP60 通过激活 IKKβ/SNAP23 轴介导的阿尔茨海默病自噬体-溶酶体融合抑制神经损伤","authors":"Wei Wang, Jun Min, Qinghua Luo, Xunhu Gu, Min Li, Xu Liu","doi":"10.1111/cns.70095","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Objective</h3>\n \n <p>The hyperphosphorylation of Tau protein is considered an important cause of neuronal degeneration in Alzheimer's disease (AD). The disruption of neuronal histone acetylation homeostasis mediated by Tip60 HAT is a common early event in neurodegenerative diseases, but the deeper regulatory mechanism on β-amyloid peptide (Aβ)-induced neurotoxicity and autophagic function in AD is still unclear.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>AD models were established both in APP/PS1 mice and Aβ<sub>1–42</sub>-treated SH-SY5Y cells. The Morris water maze test (MWM) was performed to examine mouse cognitive function. Neurological damage in the hippocampus was observed by hematoxylin–eosin (H&E), Nissl's, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and NeuN staining. Autophagosome-lysosome fusion was detected by immunohistochemistry, immunofluorescence, and Lyso-Tracker Red staining. Cell viability and apoptosis were evaluated by CCK-8 assay and flow cytometry. The molecular interactions were verified by co-immunoprecipitation (Co-IP), dual luciferase assays, and ChIP detections. The RNA and autophagy-lysosome-related proteins were assessed by Western blot and RT-qPCR.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>TIP60 overexpression improved cognitive deficits and neurological damage and restored the impairment of autophagy-lysosomes fusion in vivo. Similarly, the upregulation of TIP60 in Aβ<sub>1–42</sub>-treated SH-SY5Y cells suppressed neuronal apoptosis and tau phosphorylation through the activating autophagy-lysosome pathway. Mechanistically, TIP60 activated IKKβ transcription by promoting SOX4 acetylation, thus leading to the translocation of SNAP23 to STX17-contained autophagosomes. Moreover, the protective roles of TIP60 in neuron damage were abolished by the inhibition of SOX4/IKKβ signaling.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>Collectively, our results highlighted the potential of the TIP60 target for AD and provided new insights into the mechanisms underlying neuroprotection in this disorder.</p>\n </section>\n </div>","PeriodicalId":154,"journal":{"name":"CNS Neuroscience & Therapeutics","volume":"30 11","pages":""},"PeriodicalIF":5.0000,"publicationDate":"2024-11-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11537769/pdf/","citationCount":"0","resultStr":"{\"title\":\"Lysine Acetyltransferase TIP60 Restricts Nerve Injury by Activating IKKβ/SNAP23 Axis-Mediated Autophagosome-Lysosome Fusion in Alzheimer's Disease\",\"authors\":\"Wei Wang, Jun Min, Qinghua Luo, Xunhu Gu, Min Li, Xu Liu\",\"doi\":\"10.1111/cns.70095\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Objective</h3>\\n \\n <p>The hyperphosphorylation of Tau protein is considered an important cause of neuronal degeneration in Alzheimer's disease (AD). The disruption of neuronal histone acetylation homeostasis mediated by Tip60 HAT is a common early event in neurodegenerative diseases, but the deeper regulatory mechanism on β-amyloid peptide (Aβ)-induced neurotoxicity and autophagic function in AD is still unclear.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>AD models were established both in APP/PS1 mice and Aβ<sub>1–42</sub>-treated SH-SY5Y cells. The Morris water maze test (MWM) was performed to examine mouse cognitive function. Neurological damage in the hippocampus was observed by hematoxylin–eosin (H&E), Nissl's, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and NeuN staining. Autophagosome-lysosome fusion was detected by immunohistochemistry, immunofluorescence, and Lyso-Tracker Red staining. Cell viability and apoptosis were evaluated by CCK-8 assay and flow cytometry. The molecular interactions were verified by co-immunoprecipitation (Co-IP), dual luciferase assays, and ChIP detections. The RNA and autophagy-lysosome-related proteins were assessed by Western blot and RT-qPCR.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>TIP60 overexpression improved cognitive deficits and neurological damage and restored the impairment of autophagy-lysosomes fusion in vivo. Similarly, the upregulation of TIP60 in Aβ<sub>1–42</sub>-treated SH-SY5Y cells suppressed neuronal apoptosis and tau phosphorylation through the activating autophagy-lysosome pathway. Mechanistically, TIP60 activated IKKβ transcription by promoting SOX4 acetylation, thus leading to the translocation of SNAP23 to STX17-contained autophagosomes. Moreover, the protective roles of TIP60 in neuron damage were abolished by the inhibition of SOX4/IKKβ signaling.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusion</h3>\\n \\n <p>Collectively, our results highlighted the potential of the TIP60 target for AD and provided new insights into the mechanisms underlying neuroprotection in this disorder.</p>\\n </section>\\n </div>\",\"PeriodicalId\":154,\"journal\":{\"name\":\"CNS Neuroscience & Therapeutics\",\"volume\":\"30 11\",\"pages\":\"\"},\"PeriodicalIF\":5.0000,\"publicationDate\":\"2024-11-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11537769/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"CNS Neuroscience & Therapeutics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cns.70095\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"CNS Neuroscience & Therapeutics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cns.70095","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
目的:Tau 蛋白的过度磷酸化被认为是阿尔茨海默病(AD)神经元变性的重要原因。由Tip60 HAT介导的神经元组蛋白乙酰化平衡的破坏是神经退行性疾病中常见的早期事件,但AD中β-淀粉样肽(Aβ)诱导的神经毒性和自噬功能的深层调控机制仍不清楚:方法:在APP/PS1小鼠和Aβ1-42处理的SH-SY5Y细胞中建立AD模型。方法:在APP/PS1小鼠和Aβ1-42处理过的SH-SY5Y细胞中建立了AD模型。通过苏木精-伊红(H&E)、Nissl's、末端脱氧核苷酸转移酶DUTP缺口标记(TUNEL)和NeuN染色观察海马的神经损伤。通过免疫组化、免疫荧光和溶菌酶追踪红染色检测自噬体-溶酶体融合。细胞活力和细胞凋亡通过 CCK-8 检测法和流式细胞术进行评估。分子相互作用通过共免疫共沉淀(Co-IP)、双荧光素酶测定和 ChIP 检测进行验证。通过 Western 印迹和 RT-qPCR 评估了 RNA 和自噬溶酶体相关蛋白:结果:TIP60的过表达改善了认知障碍和神经损伤,并恢复了体内自噬-溶酶体融合的损伤。同样,在 Aβ1-42 处理的 SH-SY5Y 细胞中,TIP60 的上调通过激活自噬-溶酶体途径抑制了神经元凋亡和 tau 磷酸化。从机制上讲,TIP60通过促进SOX4乙酰化激活IKKβ转录,从而导致SNAP23转位到含有STX17的自噬体。此外,抑制SOX4/IKKβ信号传导也会取消TIP60在神经元损伤中的保护作用:总之,我们的研究结果凸显了TIP60靶点在治疗AD方面的潜力,并为这一疾病的神经保护机制提供了新的见解。
Lysine Acetyltransferase TIP60 Restricts Nerve Injury by Activating IKKβ/SNAP23 Axis-Mediated Autophagosome-Lysosome Fusion in Alzheimer's Disease
Objective
The hyperphosphorylation of Tau protein is considered an important cause of neuronal degeneration in Alzheimer's disease (AD). The disruption of neuronal histone acetylation homeostasis mediated by Tip60 HAT is a common early event in neurodegenerative diseases, but the deeper regulatory mechanism on β-amyloid peptide (Aβ)-induced neurotoxicity and autophagic function in AD is still unclear.
Methods
AD models were established both in APP/PS1 mice and Aβ1–42-treated SH-SY5Y cells. The Morris water maze test (MWM) was performed to examine mouse cognitive function. Neurological damage in the hippocampus was observed by hematoxylin–eosin (H&E), Nissl's, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and NeuN staining. Autophagosome-lysosome fusion was detected by immunohistochemistry, immunofluorescence, and Lyso-Tracker Red staining. Cell viability and apoptosis were evaluated by CCK-8 assay and flow cytometry. The molecular interactions were verified by co-immunoprecipitation (Co-IP), dual luciferase assays, and ChIP detections. The RNA and autophagy-lysosome-related proteins were assessed by Western blot and RT-qPCR.
Results
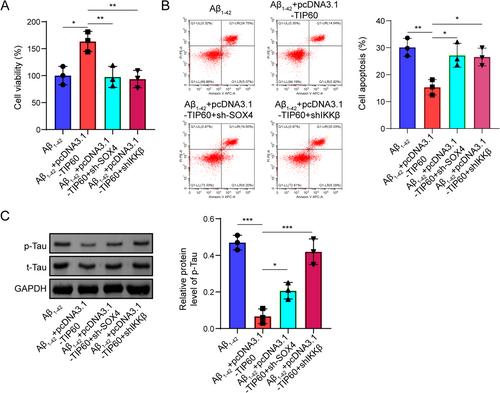
TIP60 overexpression improved cognitive deficits and neurological damage and restored the impairment of autophagy-lysosomes fusion in vivo. Similarly, the upregulation of TIP60 in Aβ1–42-treated SH-SY5Y cells suppressed neuronal apoptosis and tau phosphorylation through the activating autophagy-lysosome pathway. Mechanistically, TIP60 activated IKKβ transcription by promoting SOX4 acetylation, thus leading to the translocation of SNAP23 to STX17-contained autophagosomes. Moreover, the protective roles of TIP60 in neuron damage were abolished by the inhibition of SOX4/IKKβ signaling.
Conclusion
Collectively, our results highlighted the potential of the TIP60 target for AD and provided new insights into the mechanisms underlying neuroprotection in this disorder.
期刊介绍:
CNS Neuroscience & Therapeutics provides a medium for rapid publication of original clinical, experimental, and translational research papers, timely reviews and reports of novel findings of therapeutic relevance to the central nervous system, as well as papers related to clinical pharmacology, drug development and novel methodologies for drug evaluation. The journal focuses on neurological and psychiatric diseases such as stroke, Parkinson’s disease, Alzheimer’s disease, depression, schizophrenia, epilepsy, and drug abuse.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


