Junben Weng, Hongqiang Cui, Da Zheng, Zhenhao Zhou, Dinglin Zhang, Huiying Chu, Anhui Wang, Guohui Li
{"title":"基于多极的碳氢化合物反应力场","authors":"Junben Weng, Hongqiang Cui, Da Zheng, Zhenhao Zhou, Dinglin Zhang, Huiying Chu, Anhui Wang, Guohui Li","doi":"10.1021/acs.jctc.4c01285","DOIUrl":null,"url":null,"abstract":"<p><p>The computational complexity of quantum chemistry methods has prompted the development of reactive force fields, facilitating practical applications of molecular dynamics simulations for large-scale reactive systems. Current reactive force fields typically employ intricate corrections based on prior chemical knowledge, which severely impedes their further advancement. This study presents a new atomic multipole-based reactive model with bond free (OPERATOR). The force field is constructed on a simple, physically motivated model within the AMOEBA framework that closely resembles the physical representation of the chemical reaction processes. In the force field, the atomic multipoles are generated dynamically according to the atomic environments, aiming to effectively capture significant changes in the electrostatic environments during chemical reactions. Subsequently, atomic multipole-based charge penetration, polarization, and charge transfer effects are incorporated into the force field to describe the complex electrostatic interactions in the system. The force field also includes van der Waals interactions and three-body potentials. In addition, to extend these nonreactive interactions to chemical reactions, the atom distribution multipole moments are used to characterize different chemical environments. The force field has been optimized using the dataset of potential energy surfaces (PESs) of hydrocarbons derived from DFT results of millions of conformations with six degrees of freedom (DOFs). The results demonstrate that the new force field effectively replicates both the monopoles and the energies. In comparison to ReaxFF, the new force field exhibits comparable or superior performance. Furthermore, molecular dynamics simulations of <i>n</i>-heptane decomposition effectively reproduce the primary products and reactions observed in the experiments. Given the simplicity and physically motivated nature of the model, it is expected that the new force field will be utilized in future studies to investigate chemical reaction mechanisms involving more elements.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"10045-10058"},"PeriodicalIF":5.5000,"publicationDate":"2024-11-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A Multipole-Based Reactive Force Field for Hydrocarbons.\",\"authors\":\"Junben Weng, Hongqiang Cui, Da Zheng, Zhenhao Zhou, Dinglin Zhang, Huiying Chu, Anhui Wang, Guohui Li\",\"doi\":\"10.1021/acs.jctc.4c01285\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The computational complexity of quantum chemistry methods has prompted the development of reactive force fields, facilitating practical applications of molecular dynamics simulations for large-scale reactive systems. Current reactive force fields typically employ intricate corrections based on prior chemical knowledge, which severely impedes their further advancement. This study presents a new atomic multipole-based reactive model with bond free (OPERATOR). The force field is constructed on a simple, physically motivated model within the AMOEBA framework that closely resembles the physical representation of the chemical reaction processes. In the force field, the atomic multipoles are generated dynamically according to the atomic environments, aiming to effectively capture significant changes in the electrostatic environments during chemical reactions. Subsequently, atomic multipole-based charge penetration, polarization, and charge transfer effects are incorporated into the force field to describe the complex electrostatic interactions in the system. The force field also includes van der Waals interactions and three-body potentials. In addition, to extend these nonreactive interactions to chemical reactions, the atom distribution multipole moments are used to characterize different chemical environments. The force field has been optimized using the dataset of potential energy surfaces (PESs) of hydrocarbons derived from DFT results of millions of conformations with six degrees of freedom (DOFs). The results demonstrate that the new force field effectively replicates both the monopoles and the energies. In comparison to ReaxFF, the new force field exhibits comparable or superior performance. Furthermore, molecular dynamics simulations of <i>n</i>-heptane decomposition effectively reproduce the primary products and reactions observed in the experiments. Given the simplicity and physically motivated nature of the model, it is expected that the new force field will be utilized in future studies to investigate chemical reaction mechanisms involving more elements.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"10045-10058\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-11-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c01285\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01285","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/4 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
A Multipole-Based Reactive Force Field for Hydrocarbons.
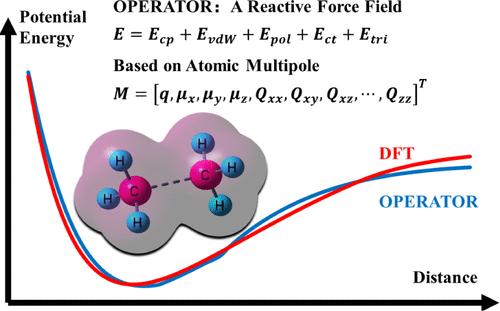
The computational complexity of quantum chemistry methods has prompted the development of reactive force fields, facilitating practical applications of molecular dynamics simulations for large-scale reactive systems. Current reactive force fields typically employ intricate corrections based on prior chemical knowledge, which severely impedes their further advancement. This study presents a new atomic multipole-based reactive model with bond free (OPERATOR). The force field is constructed on a simple, physically motivated model within the AMOEBA framework that closely resembles the physical representation of the chemical reaction processes. In the force field, the atomic multipoles are generated dynamically according to the atomic environments, aiming to effectively capture significant changes in the electrostatic environments during chemical reactions. Subsequently, atomic multipole-based charge penetration, polarization, and charge transfer effects are incorporated into the force field to describe the complex electrostatic interactions in the system. The force field also includes van der Waals interactions and three-body potentials. In addition, to extend these nonreactive interactions to chemical reactions, the atom distribution multipole moments are used to characterize different chemical environments. The force field has been optimized using the dataset of potential energy surfaces (PESs) of hydrocarbons derived from DFT results of millions of conformations with six degrees of freedom (DOFs). The results demonstrate that the new force field effectively replicates both the monopoles and the energies. In comparison to ReaxFF, the new force field exhibits comparable or superior performance. Furthermore, molecular dynamics simulations of n-heptane decomposition effectively reproduce the primary products and reactions observed in the experiments. Given the simplicity and physically motivated nature of the model, it is expected that the new force field will be utilized in future studies to investigate chemical reaction mechanisms involving more elements.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


