{"title":"通过使用不同的色谱方法和生物信息学管道优化蛋白质鉴定。","authors":"Jesus D. Castaño, Francis Beaudry","doi":"10.1002/rcm.9937","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n <h3> Rationale</h3>\n \n <p>Selection of proteomic workflows for a given project can be a daunting task. This research provides a guide outlining the impact on protein identification of different steps such as chromatographic separation, data acquisition strategies, and bioinformatic pipelines. The data presented here will help experts and nonexpert proteomic users to increase proteome coverage and peptide identification.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>HeLa protein digests were analyzed through different C18 chromatographic columns (15 and 50 cm in length), using top 12 data-dependent acquisition (DDA), top 20 DDA, and data-independent acquisition (DIA) with a nanospray source in positive mode in a Thermo Q Exactive instrument. The raw data were analyzed using different search engines, rescoring approaches, and multi-engine searches. The results were analyzed in the context of peptide and protein identifications, precursor properties, and computation requirements to understand the differences between methods.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Our results showed that higher column lengths and top <i>N</i> DDA approaches were able to significantly increase protein identifications. The use of multiple search engines yielded limited gains, whereas the use of rescoring methods clearly outperformed other strategies. Finally, DIA approaches, although successful at generating new identifications, had a limited performance influenced by the previous collection of DDA data, which could prohibitively increase instrument time. Nonetheless, the use of library-free methods showed promising results.</p>\n </section>\n \n <section>\n \n <h3> Conclusions</h3>\n \n <p>Our results highlight the impact of different experimental approaches on proteome coverage. Changes in chromatographic columns, data acquisition, or bioinformatic analysis can significantly increase the number of protein identifications (>400%). Thus, this research provides a reference upon which to build a successful proteomic workflow with different considerations at every step.</p>\n </section>\n </div>","PeriodicalId":225,"journal":{"name":"Rapid Communications in Mass Spectrometry","volume":"39 1","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-11-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/rcm.9937","citationCount":"0","resultStr":"{\"title\":\"Optimization of protein identifications through the use of different chromatographic approaches and bioinformatic pipelines\",\"authors\":\"Jesus D. Castaño, Francis Beaudry\",\"doi\":\"10.1002/rcm.9937\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <section>\\n \\n <h3> Rationale</h3>\\n \\n <p>Selection of proteomic workflows for a given project can be a daunting task. This research provides a guide outlining the impact on protein identification of different steps such as chromatographic separation, data acquisition strategies, and bioinformatic pipelines. The data presented here will help experts and nonexpert proteomic users to increase proteome coverage and peptide identification.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>HeLa protein digests were analyzed through different C18 chromatographic columns (15 and 50 cm in length), using top 12 data-dependent acquisition (DDA), top 20 DDA, and data-independent acquisition (DIA) with a nanospray source in positive mode in a Thermo Q Exactive instrument. The raw data were analyzed using different search engines, rescoring approaches, and multi-engine searches. The results were analyzed in the context of peptide and protein identifications, precursor properties, and computation requirements to understand the differences between methods.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Our results showed that higher column lengths and top <i>N</i> DDA approaches were able to significantly increase protein identifications. The use of multiple search engines yielded limited gains, whereas the use of rescoring methods clearly outperformed other strategies. Finally, DIA approaches, although successful at generating new identifications, had a limited performance influenced by the previous collection of DDA data, which could prohibitively increase instrument time. Nonetheless, the use of library-free methods showed promising results.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusions</h3>\\n \\n <p>Our results highlight the impact of different experimental approaches on proteome coverage. Changes in chromatographic columns, data acquisition, or bioinformatic analysis can significantly increase the number of protein identifications (>400%). Thus, this research provides a reference upon which to build a successful proteomic workflow with different considerations at every step.</p>\\n </section>\\n </div>\",\"PeriodicalId\":225,\"journal\":{\"name\":\"Rapid Communications in Mass Spectrometry\",\"volume\":\"39 1\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-11-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/rcm.9937\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Rapid Communications in Mass Spectrometry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/rcm.9937\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rapid Communications in Mass Spectrometry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/rcm.9937","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Optimization of protein identifications through the use of different chromatographic approaches and bioinformatic pipelines
Rationale
Selection of proteomic workflows for a given project can be a daunting task. This research provides a guide outlining the impact on protein identification of different steps such as chromatographic separation, data acquisition strategies, and bioinformatic pipelines. The data presented here will help experts and nonexpert proteomic users to increase proteome coverage and peptide identification.
Methods
HeLa protein digests were analyzed through different C18 chromatographic columns (15 and 50 cm in length), using top 12 data-dependent acquisition (DDA), top 20 DDA, and data-independent acquisition (DIA) with a nanospray source in positive mode in a Thermo Q Exactive instrument. The raw data were analyzed using different search engines, rescoring approaches, and multi-engine searches. The results were analyzed in the context of peptide and protein identifications, precursor properties, and computation requirements to understand the differences between methods.
Results
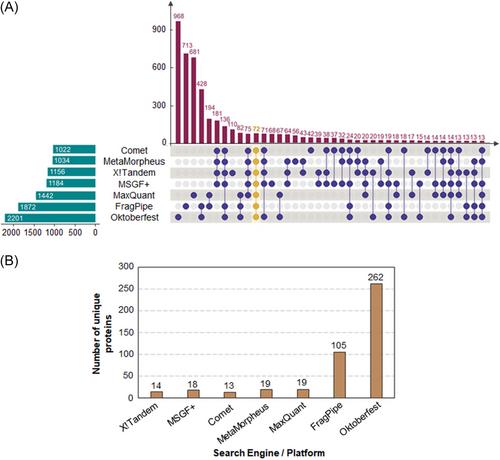
Our results showed that higher column lengths and top N DDA approaches were able to significantly increase protein identifications. The use of multiple search engines yielded limited gains, whereas the use of rescoring methods clearly outperformed other strategies. Finally, DIA approaches, although successful at generating new identifications, had a limited performance influenced by the previous collection of DDA data, which could prohibitively increase instrument time. Nonetheless, the use of library-free methods showed promising results.
Conclusions
Our results highlight the impact of different experimental approaches on proteome coverage. Changes in chromatographic columns, data acquisition, or bioinformatic analysis can significantly increase the number of protein identifications (>400%). Thus, this research provides a reference upon which to build a successful proteomic workflow with different considerations at every step.
期刊介绍:
Rapid Communications in Mass Spectrometry is a journal whose aim is the rapid publication of original research results and ideas on all aspects of the science of gas-phase ions; it covers all the associated scientific disciplines. There is no formal limit on paper length ("rapid" is not synonymous with "brief"), but papers should be of a length that is commensurate with the importance and complexity of the results being reported. Contributions may be theoretical or practical in nature; they may deal with methods, techniques and applications, or with the interpretation of results; they may cover any area in science that depends directly on measurements made upon gaseous ions or that is associated with such measurements.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


