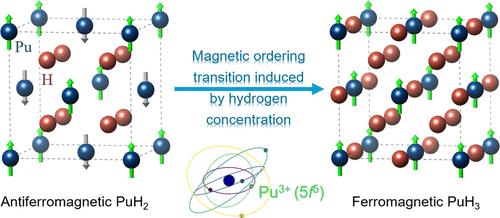
{"title":"钚氢化物的 DFT + U 研究与占位矩阵控制。","authors":"Liuhua Xie, Xiaoqiu Ye, Ruizhi Qiu","doi":"10.1021/acs.inorgchem.4c02823","DOIUrl":null,"url":null,"abstract":"<p><p>The magnetic order of plutonium hydrides (PuH<sub><i>x</i></sub>) has long been a subject of controversy, both experimentally and theoretically. The magnetic, structural, electronic, and thermodynamic properties of PuH<sub><i>x</i></sub> are investigated using Hubbard-corrected density functional theory (DFT + <i>U</i>), with <i>U</i> derived from linear response calculations. To address the issue of electronic metastable states within DFT + <i>U</i>, we employ an occupation matrix control method as well as allowing 5f orbitals to break the structural symmetry. This advanced computational approach establishes that the ground-state magnetic order for PuH<sub>2</sub> is antiferromagnetic and that for PuH<sub>3</sub> is ferromagnetic, consistent with Faraday and nuclear magnetic resonance experiments. Using the conventional hydrogen-vacancy model for PuH<sub><i>x</i></sub>, the hydrogen-content-induced magnetic transition and anomalous variation of the magnetic moment are successfully reproduced. In particular, the calculated transition point of <i>x</i> matches the experimental findings. Furthermore, the electronic configuration of the Pu atom in PuH<sub><i>x</i></sub> is determined to be 5f<sup>5</sup>, which is consistent with observations from X-ray photoemission spectroscopy. Our calculated values for enthalpy of formation, heat capacity, and entropy are in good agreement with experimental data, thereby validating the robustness of our theoretical framework.</p>","PeriodicalId":40,"journal":{"name":"Inorganic Chemistry","volume":" ","pages":"21885-21897"},"PeriodicalIF":4.7000,"publicationDate":"2024-11-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"DFT + <i>U</i> Study of Plutonium Hydrides with Occupation Matrix Control.\",\"authors\":\"Liuhua Xie, Xiaoqiu Ye, Ruizhi Qiu\",\"doi\":\"10.1021/acs.inorgchem.4c02823\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The magnetic order of plutonium hydrides (PuH<sub><i>x</i></sub>) has long been a subject of controversy, both experimentally and theoretically. The magnetic, structural, electronic, and thermodynamic properties of PuH<sub><i>x</i></sub> are investigated using Hubbard-corrected density functional theory (DFT + <i>U</i>), with <i>U</i> derived from linear response calculations. To address the issue of electronic metastable states within DFT + <i>U</i>, we employ an occupation matrix control method as well as allowing 5f orbitals to break the structural symmetry. This advanced computational approach establishes that the ground-state magnetic order for PuH<sub>2</sub> is antiferromagnetic and that for PuH<sub>3</sub> is ferromagnetic, consistent with Faraday and nuclear magnetic resonance experiments. Using the conventional hydrogen-vacancy model for PuH<sub><i>x</i></sub>, the hydrogen-content-induced magnetic transition and anomalous variation of the magnetic moment are successfully reproduced. In particular, the calculated transition point of <i>x</i> matches the experimental findings. Furthermore, the electronic configuration of the Pu atom in PuH<sub><i>x</i></sub> is determined to be 5f<sup>5</sup>, which is consistent with observations from X-ray photoemission spectroscopy. Our calculated values for enthalpy of formation, heat capacity, and entropy are in good agreement with experimental data, thereby validating the robustness of our theoretical framework.</p>\",\"PeriodicalId\":40,\"journal\":{\"name\":\"Inorganic Chemistry\",\"volume\":\" \",\"pages\":\"21885-21897\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-11-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Inorganic Chemistry\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.inorgchem.4c02823\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganic Chemistry","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1021/acs.inorgchem.4c02823","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
摘要
长期以来,钚氢化物(PuHx)的磁序一直是实验和理论上存在争议的问题。我们使用哈伯德校正密度泛函理论(DFT + U)研究了 PuHx 的磁性、结构、电子和热力学性质,其中 U 来自线性响应计算。为了解决 DFT + U 中的电子陨变态问题,我们采用了占位矩阵控制方法,并允许 5f 轨道打破结构对称性。这种先进的计算方法确定了 PuH2 的基态磁序是反铁磁的,而 PuH3 的基态磁序是铁磁的,这与法拉第和核磁共振实验相一致。利用 PuHx 的传统氢空位模型,成功地再现了氢含量诱导的磁转变和磁矩的异常变化。特别是,x 的计算转变点与实验结果相吻合。此外,还确定了 PuHx 中 Pu 原子的电子构型为 5f5,这与 X 射线光发射光谱的观测结果一致。我们计算出的形成焓、热容量和熵值与实验数据十分吻合,从而验证了我们理论框架的稳健性。
DFT + U Study of Plutonium Hydrides with Occupation Matrix Control.
The magnetic order of plutonium hydrides (PuHx) has long been a subject of controversy, both experimentally and theoretically. The magnetic, structural, electronic, and thermodynamic properties of PuHx are investigated using Hubbard-corrected density functional theory (DFT + U), with U derived from linear response calculations. To address the issue of electronic metastable states within DFT + U, we employ an occupation matrix control method as well as allowing 5f orbitals to break the structural symmetry. This advanced computational approach establishes that the ground-state magnetic order for PuH2 is antiferromagnetic and that for PuH3 is ferromagnetic, consistent with Faraday and nuclear magnetic resonance experiments. Using the conventional hydrogen-vacancy model for PuHx, the hydrogen-content-induced magnetic transition and anomalous variation of the magnetic moment are successfully reproduced. In particular, the calculated transition point of x matches the experimental findings. Furthermore, the electronic configuration of the Pu atom in PuHx is determined to be 5f5, which is consistent with observations from X-ray photoemission spectroscopy. Our calculated values for enthalpy of formation, heat capacity, and entropy are in good agreement with experimental data, thereby validating the robustness of our theoretical framework.
期刊介绍:
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry. Coverage includes experimental and theoretical reports on quantitative studies of structure and thermodynamics, kinetics, mechanisms of inorganic reactions, bioinorganic chemistry, and relevant aspects of organometallic chemistry, solid-state phenomena, and chemical bonding theory. Emphasis is placed on the synthesis, structure, thermodynamics, reactivity, spectroscopy, and bonding properties of significant new and known compounds.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


