降低成本计算和利用无障碍过程的精确径向相互作用势能
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
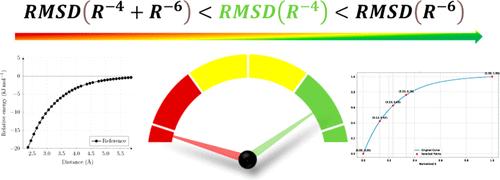
无障碍步骤是气相中自由基和离子反应的典型特征,这些反应通常发生在极端环境中,如在极高温下运行的燃烧反应器或以超低温和超低压为特征的星际介质。在模拟这些环境的条件下进行实验研究非常困难,这表明计算方法可以提供宝贵的支持。在这方面,过渡态理论框架下对这些过程的最先进处理方法能够提供精确的动力学参数,前提是底层势能面足够精确。由于这需要平衡处理静态和动态相关性(在不同区域发挥不同作用),因此需要非常复杂和昂贵的量子化学方法。解决这一问题的有效方法之一是计算精确的一维径向势,然后用它来校正用更便宜的量子化学方法进行的蒙特卡罗采样结果。在本文中,我们将证明,对于大量不同的无障碍反应步骤,径向位势是由 R-4 项决定的,再加上 R-6 项的贡献,就能与参考点达成定量一致。这一结果的后果并非微不足道,因为参考势能只能由数量非常有限的点拟合,这些点的间距可能是非线性的。对于由长程过渡态控制的反应步骤,还给出了在相空间理论框架内计算反应速率的通用表达式。所有这些改进都为计算涉及大分子的无障碍反应的反应速率铺平了道路。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Reduced Cost Computation and Exploitation of Accurate Radial Interaction Potentials for Barrier-Less Processes
Barrier-less steps are typical of radical and ionic reactions in the gas-phase, which often take place in extreme environments such as the combustion reactors operating at very high temperatures or the interstellar medium, characterized by ultralow temperatures and pressures. The difficulty of experimental studies in conditions mimicking these environments suggests that computational approaches can provide a valuable support. In this connection, the most advanced treatments of these processes in the framework of transition state theory are able to deliver accurate kinetic parameters provided that the underlying potential energy surface is sufficiently accurate. Since this requires a balanced treatment of static and dynamic correlation (which play different roles in different regions), very sophisticated and expensive quantum chemical approaches are required. One effective solution of this problem is offered by the computation of accurate one-dimensional radial potentials, which are then used to correct the results of a Monte Carlo sampling performed by cheaper quantum chemical approaches. In this paper, we will show that, for a large panel of different barrier-less reaction steps, the radial potential is ruled by the R–4 term and that addition of a further R–6 contribution provides quantitative agreement with the reference points. The consequences of this outcome are not trivial, since the reference potential can be fitted by a very limited number of points possibly with a nonlinear spacing. In the case of reaction steps ruled by long-range transition states, generalized expressions are also given for computing reaction rates in the framework of the phase-space theory. All these improvements pave the way toward the computation of reaction rates for barrier-less reactions involving large molecules.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


