pH 值介导的溶液相质子转移推动了碱性电解质中苯酚的强化电化学氢化反应
IF 11.3
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
苯酚和其他生物质衍生模型化合物的电化学氢化(ECH)的远达效率(FE)有可能通过在碱性电解质中操作而得到改善,在碱性电解质中,由于 Volmer 阶跃势垒较高,寄生氢演化反应速率明显较慢。然而,这种方法可能会受到吸附氢(Had)形成的较高障碍的影响,因为碳氢化合物的 ECH 预计会受到氢原子转移的限制,并通过 Langmuir-Hinshelwood 型(LH)机制进行。在这项研究中,我们发现铂和铑这两种常见的 ECH 反应催化剂在苯酚 ECH 反应中的 pH 值变化趋势截然不同。在 pH ≤ 5 的酸性电解质中,铂上的苯酚 ECH FE 和速率最高,而在 pH 9-10 附近,Rh 上的活性最高。虽然我们的动力学分析支持铂在所有 pH 值下的 LH 机制,但苯酚在 Rh 上的 ECH 从低 pH 值下的 LH 机制转变为直接质子耦合电子转移(Eley-Rideal 型机制),其中表面吸附的苯酚通过溶液相 H 转移进行氢化。我们发现,由于 pH 值接近苯酚的 pKa(pKa = 10.0),因此 Rh 在 pH 值为 9-10 时会出现活性峰值。当电解质的 pH 值与其 pKa 值相匹配时,苯酚质子化/去质子化的可逆性有助于介导溶液中的氢转移到吸附的苯酚上。我们还讨论了缓冲物种在减轻局部 pH 值变化方面的作用,以及在碱性 pH 值下苯酚在 Rh 上的 ECH 中作为 H 供体的作用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
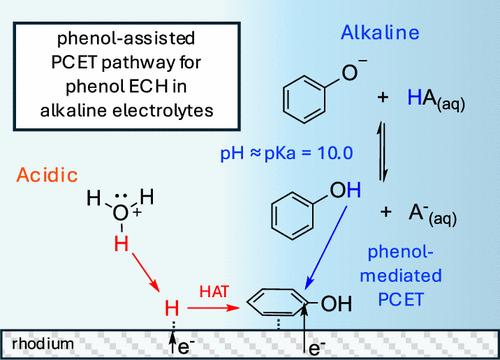
pH-Mediated Solution-Phase Proton Transfer Drives Enhanced Electrochemical Hydrogenation of Phenol in Alkaline Electrolyte
The faradaic efficiency (FE) of the electrochemical hydrogenation (ECH) of phenol and other biomass-derived model compounds could potentially be improved by operating in alkaline electrolytes, where the parasitic hydrogen evolution reaction rate is significantly slower due to a higher Volmer step barrier. However, this approach is potentially limited by the impact of the higher barrier for adsorbed hydrogen (Had) formation, as hydrocarbon ECH is expected to be limited by a hydrogen atom transfer, progressing through a Langmuir–Hinshelwood-type (LH) mechanism. In this work, we show that there are contrasting pH trends for phenol ECH between Pt and Rh, two common catalysts for ECH reactions. Phenol ECH FE and rate on Pt is highest in acidic electrolytes of pH ≤ 5, while activity on Rh is highest near pH 9–10. While our kinetic analysis supports a LH mechanism for Pt at all pH, phenol ECH on Rh shifts from a LH mechanism at low pH to being limited by a direct proton-coupled electron transfer (Eley–Rideal-type mechanism) in which surface adsorbed phenol is hydrogenated by solution-phase H-transfer. We show that the peak activity on Rh at pH 9–10 is due to the proximity of the pH to the pKa of phenol (pKa = 10.0). The reversibility of protonation/deprotonation of phenol when electrolyte pH matches its pKa helps to mediate H-transfer from solution to adsorbed phenol. We also discuss the role of buffer species in mitigating the local pH change and as a H-donor in phenol ECH on Rh at alkaline pH.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


