通过可解释的几何深度学习,GeoNet 可准确预测蛋白质配体结合位点
IF 4.4
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
蛋白质结合残基的鉴定对于了解其体内功能至关重要。然而,由于缺乏已知的残基结合模式,准确识别结合位点仍然是一项计算挑战。残基的局部空间分布及其交互式生物物理环境都决定了结合模式。以前的方法无法同时捕获这两种信息,导致性能不尽人意。在此,我们介绍一种可解释的几何深度学习模型--GeoNet,该模型通过学习潜在的残基结合模式来预测 DNA、RNA 和蛋白质的结合位点。为此,GeoNet 引入了一种无坐标几何表示法来描述局部残基分布,并生成一个特征空间来描绘局部交互式生物物理环境。评估结果表明,GeoNet 优于其他领先的预测工具,并显示了所学表征的强大可解释性。我们介绍了三个测试案例,其中使用 GeoNet 成功识别了交互界面。本文章由计算机程序翻译,如有差异,请以英文原文为准。
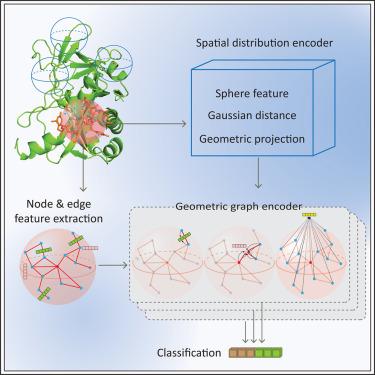
GeoNet enables the accurate prediction of protein-ligand binding sites through interpretable geometric deep learning
The identification of protein binding residues is essential for understanding their functions in vivo. However, it remains a computational challenge to accurately identify binding sites due to the lack of known residue binding patterns. Local residue spatial distribution and its interactive biophysical environment both determine binding patterns. Previous methods could not capture both information simultaneously, resulting in unsatisfactory performance. Here, we present GeoNet, an interpretable geometric deep learning model for predicting DNA, RNA, and protein binding sites by learning the latent residue binding patterns. GeoNet achieves this by introducing a coordinate-free geometric representation to characterize local residue distributions and generating an eigenspace to depict local interactive biophysical environments. Evaluation shows that GeoNet is superior compared to other leading predictors and it shows a strong interpretability of learned representations. We present three test cases, where interaction interfaces were successfully identified with GeoNet.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Structure
生物-生化与分子生物学
CiteScore
8.90
自引率
1.80%
发文量
155
审稿时长
3-8 weeks
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


