了解过渡金属氧化物电化学氧化二氮的活性趋势
IF 11.3
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
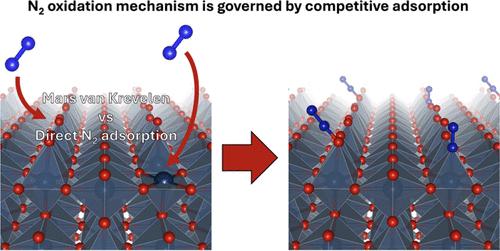
硝酸(HNO3)是一种重要的商品化学品,通过奥斯特瓦尔德过程中氨 NH3 的氧化作用大规模生产,因此在全球温室气体排放中占很大比例。通过电化学氮氧化反应(N2OR)直接氧化二氮生成硝酸是一种极具吸引力的替代方法,但迄今为止,这种方法在很大程度上仍然难以实现。为了从根本上了解 N2OR 的局限性,我们在本文中研究了氮(N2)和氢氧化物(OH)等水氧化中间产物在一系列过渡金属氧化物上的竞争吸附动力学。利用密度泛函理论(DFT)计算,我们探索了三种可能的 N2OR 机制:N2 的直接吸附和解离吸附,以及涉及 N2 吸附在表面结合的原子氧上的 Mars-van Krevelen(MvK)型机制。我们观察到 N2 和 OH 在金属端过渡金属氧化物上的吸附能之间存在很强的线性比例关系,这表明在 N2OR 的典型高氧化工作条件下(URHE > 1.24 V),水氧化中间产物(如 OH)很可能在表面占主导地位,从而导致吸附 N2 的覆盖范围极小。根据这一结果,我们发现直接吸附或解离吸附 N2 的可能性不大,这表明 N2OR 存在 MvK 型机制。通过使用 DFT 对该机制进行进一步探究,我们发现由于 N2 吸附的活化势垒较高,该反应的能效在很大程度上低于水氧化,而我们发现活化势垒是该过程的速率决定步骤。我们的实验结果证实了这些发现,大多数测试催化剂都表现出较低的 N2OR 选择性和涉及 N2(g)的速率决定步骤。然而,动态电位控制可能会限制氧进化反应(OER)并促进 N2 吸附,因此成为提高 N2OR 活性的一种可能策略。这项工作强调了实现高效 N2OR 所面临的挑战,突出了非传统催化剂设计和操作策略的必要性,如电解质工程和动态电位控制,以克服固有的动力学和热力学障碍。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Understanding Activity Trends in Electrochemical Dinitrogen Oxidation over Transition Metal Oxides
Nitric acid (HNO3) is a critical commodity chemical produced on an enormous scale via oxidation of ammonia NH3 in the Ostwald process and, as such, is responsible for a significant fraction of global greenhouse gas emissions. Formation of nitric acid by direct oxidation of dinitrogen via the electrochemical nitrogen oxidation reaction (N2OR) is an attractive alternative but has so far largely remained elusive. Toward advancing our fundamental understanding of the limitations of the N2OR, in this article, we investigated the competitive adsorption dynamics of nitrogen (N2) and water oxidation intermediates such as hydroxide (OH) on a range of transition metal oxides. Using density functional theory (DFT) calculations, we explore three possible N2OR mechanisms: direct adsorption and dissociative adsorption of N2, and a Mars-van Krevelen (MvK)-type mechanism involving the adsorption of N2 on a surface-bound atomic oxygen. We observed a strong linear scaling relation between the adsorption energy of N2 and OH on the metal-terminated transition metal oxide, suggesting that under typical highly oxidizing operating conditions for the N2OR (URHE > 1.24 V), water oxidation intermediates such as OH are likely to dominate the surface, leading to vanishingly small coverage of adsorbed N2. From this result, we find that direct or dissociative adsorption of N2 is unlikely, suggesting an MvK-type mechanism for the N2OR. Probing this mechanism further using DFT, we find that the reaction energetics are largely less favorable than water oxidation due to the high activation barrier for N2 adsorption, which we find to be the rate-determining step for the process. Our experimental results corroborate these findings with the majority of tested catalysts exhibiting poor N2OR selectivity and a rate-determining step involving N2(g). However, dynamic potential control emerged as a possible strategy to enhance N2OR activity as it may limit the oxygen evolution reaction (OER) and promote N2 adsorption. This work underscores the challenges in achieving efficient N2OR, highlighting the need for unconventional catalyst designs and operational strategies, such as electrolyte engineering and dynamic potential control, to overcome the inherent kinetic and thermodynamic barriers.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


