Jun Li, Feierkaiti Yushanjiang, Zhao Fang, Wan-li Liu
{"title":"NAT10 介导的 RNA ac4C 乙酰化有助于心肌梗死诱发的心脏纤维化。","authors":"Jun Li, Feierkaiti Yushanjiang, Zhao Fang, Wan-li Liu","doi":"10.1111/jcmm.70141","DOIUrl":null,"url":null,"abstract":"<p>Cardiac fibrosis is featured cardiac fibroblast activation and extracellular matrix accumulation. Ac4C acetylation is an important epigenetic regulation of RNAs that has been recently discovered, and it is solely carried out by NAT10, the exclusive enzyme used for the modification. However, the potential regulatory mechanisms of ac4C acetylation in myocardial fibrosis following myocardial infarction remain poorly understood. In our study, we activated fibroblasts in vitro using TGF-β1 (20 ng/mL), followed by establishing a myocardial infarction mouse model to evaluate the impact of NAT10 on collagen synthesis and cardiac fibroblast proliferation. We utilized a NAT10 inhibitor, Remodelin, to attenuate the acetylation capacity of NAT10. In the cardiac fibrosis tissues of chronic myocardial infarction mice and cultured cardiac fibroblasts (CFs) in response to TGF-β1 treatment, there was an elevation in the levels of NAT10 expression. This increase facilitated proliferation, the accumulation of collagens, as well as fibroblast-to-myofibroblast transition. Through the administration of Remodelin, we effectively reduced cardiac fibrosis in myocardial infarction mice by inhibiting NAT10's ability to acetylate mRNA. Inhibition of NAT10 resulted in changes in collagen-related gene expression and ac4C acetylation levels. Mechanistically, we found that NAT10 upregulates the acetylation modification of BCL-XL mRNA and enhances the stability of BCL-XL mRNA, thereby upregulating its protein expression, inhibiting the activation of Caspase3 and blocking the apoptosis of CFs. Therefore, the crucial involvement of NAT10-mediated ac4C acetylation is significant in the cardiac fibrosis progression, affording promising molecular targets for the treatment of fibrosis and relevant cardiac diseases.</p>","PeriodicalId":101321,"journal":{"name":"JOURNAL OF CELLULAR AND MOLECULAR MEDICINE","volume":"28 21","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jcmm.70141","citationCount":"0","resultStr":"{\"title\":\"NAT10-mediated RNA ac4C acetylation contributes to the myocardial infarction-induced cardiac fibrosis\",\"authors\":\"Jun Li, Feierkaiti Yushanjiang, Zhao Fang, Wan-li Liu\",\"doi\":\"10.1111/jcmm.70141\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Cardiac fibrosis is featured cardiac fibroblast activation and extracellular matrix accumulation. Ac4C acetylation is an important epigenetic regulation of RNAs that has been recently discovered, and it is solely carried out by NAT10, the exclusive enzyme used for the modification. However, the potential regulatory mechanisms of ac4C acetylation in myocardial fibrosis following myocardial infarction remain poorly understood. In our study, we activated fibroblasts in vitro using TGF-β1 (20 ng/mL), followed by establishing a myocardial infarction mouse model to evaluate the impact of NAT10 on collagen synthesis and cardiac fibroblast proliferation. We utilized a NAT10 inhibitor, Remodelin, to attenuate the acetylation capacity of NAT10. In the cardiac fibrosis tissues of chronic myocardial infarction mice and cultured cardiac fibroblasts (CFs) in response to TGF-β1 treatment, there was an elevation in the levels of NAT10 expression. This increase facilitated proliferation, the accumulation of collagens, as well as fibroblast-to-myofibroblast transition. Through the administration of Remodelin, we effectively reduced cardiac fibrosis in myocardial infarction mice by inhibiting NAT10's ability to acetylate mRNA. Inhibition of NAT10 resulted in changes in collagen-related gene expression and ac4C acetylation levels. Mechanistically, we found that NAT10 upregulates the acetylation modification of BCL-XL mRNA and enhances the stability of BCL-XL mRNA, thereby upregulating its protein expression, inhibiting the activation of Caspase3 and blocking the apoptosis of CFs. Therefore, the crucial involvement of NAT10-mediated ac4C acetylation is significant in the cardiac fibrosis progression, affording promising molecular targets for the treatment of fibrosis and relevant cardiac diseases.</p>\",\"PeriodicalId\":101321,\"journal\":{\"name\":\"JOURNAL OF CELLULAR AND MOLECULAR MEDICINE\",\"volume\":\"28 21\",\"pages\":\"\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jcmm.70141\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JOURNAL OF CELLULAR AND MOLECULAR MEDICINE\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/jcmm.70141\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JOURNAL OF CELLULAR AND MOLECULAR MEDICINE","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jcmm.70141","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
NAT10-mediated RNA ac4C acetylation contributes to the myocardial infarction-induced cardiac fibrosis
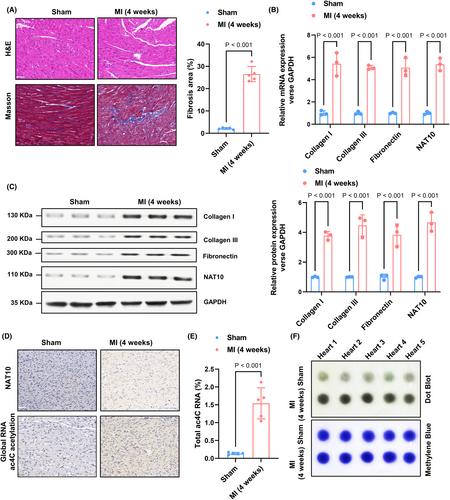
Cardiac fibrosis is featured cardiac fibroblast activation and extracellular matrix accumulation. Ac4C acetylation is an important epigenetic regulation of RNAs that has been recently discovered, and it is solely carried out by NAT10, the exclusive enzyme used for the modification. However, the potential regulatory mechanisms of ac4C acetylation in myocardial fibrosis following myocardial infarction remain poorly understood. In our study, we activated fibroblasts in vitro using TGF-β1 (20 ng/mL), followed by establishing a myocardial infarction mouse model to evaluate the impact of NAT10 on collagen synthesis and cardiac fibroblast proliferation. We utilized a NAT10 inhibitor, Remodelin, to attenuate the acetylation capacity of NAT10. In the cardiac fibrosis tissues of chronic myocardial infarction mice and cultured cardiac fibroblasts (CFs) in response to TGF-β1 treatment, there was an elevation in the levels of NAT10 expression. This increase facilitated proliferation, the accumulation of collagens, as well as fibroblast-to-myofibroblast transition. Through the administration of Remodelin, we effectively reduced cardiac fibrosis in myocardial infarction mice by inhibiting NAT10's ability to acetylate mRNA. Inhibition of NAT10 resulted in changes in collagen-related gene expression and ac4C acetylation levels. Mechanistically, we found that NAT10 upregulates the acetylation modification of BCL-XL mRNA and enhances the stability of BCL-XL mRNA, thereby upregulating its protein expression, inhibiting the activation of Caspase3 and blocking the apoptosis of CFs. Therefore, the crucial involvement of NAT10-mediated ac4C acetylation is significant in the cardiac fibrosis progression, affording promising molecular targets for the treatment of fibrosis and relevant cardiac diseases.
期刊介绍:
The Journal of Cellular and Molecular Medicine serves as a bridge between physiology and cellular medicine, as well as molecular biology and molecular therapeutics. With a 20-year history, the journal adopts an interdisciplinary approach to showcase innovative discoveries.
It publishes research aimed at advancing the collective understanding of the cellular and molecular mechanisms underlying diseases. The journal emphasizes translational studies that translate this knowledge into therapeutic strategies. Being fully open access, the journal is accessible to all readers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


