{"title":"钙电池电解质溶解结构的计算预测","authors":"Heonjae Jeong, Haimeng Wang and Lei Cheng","doi":"10.1039/D4TA06675H","DOIUrl":null,"url":null,"abstract":"<p >Calcium ion batteries are emerging as a key focus in the pursuit of alternatives to lithium-ion batteries. However, a crucial gap remains in understanding how different electrolyte species influence their solvation structures. In this study, we demonstrate a comprehensive predictive approach that integrates <em>ab initio</em> calculations and machine learning force fields (MLFFs) to address this challenge. Using <em>ab initio</em> molecular dynamics (AIMD) simulations, we accurately predict the solvation structures within the first solvation shell, while also evaluating their reductive and oxidative stability through frontier orbital analysis. This analysis compares both implicit and explicit electrolyte conditions. To further elucidate these structures, we calculate and visualize their formation free energies using density functional theory (DFT), combined with heat map analysis. Additionally, MLFF simulations extend our predictions to nanosecond-scale trajectories, surpassing the limitations of picosecond-scale AIMD. The predicted solvated structures show strong agreement with both AIMD and DFT results, demonstrating the robustness of our approach. Thus, by leveraging these comprehensive methods, we provide a more reliable framework for predicting solvation structures in calcium ion and other battery electrolytes.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 47","pages":" 33150-33161"},"PeriodicalIF":9.5000,"publicationDate":"2024-10-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational prediction of solvation structures in calcium battery electrolytes†\",\"authors\":\"Heonjae Jeong, Haimeng Wang and Lei Cheng\",\"doi\":\"10.1039/D4TA06675H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Calcium ion batteries are emerging as a key focus in the pursuit of alternatives to lithium-ion batteries. However, a crucial gap remains in understanding how different electrolyte species influence their solvation structures. In this study, we demonstrate a comprehensive predictive approach that integrates <em>ab initio</em> calculations and machine learning force fields (MLFFs) to address this challenge. Using <em>ab initio</em> molecular dynamics (AIMD) simulations, we accurately predict the solvation structures within the first solvation shell, while also evaluating their reductive and oxidative stability through frontier orbital analysis. This analysis compares both implicit and explicit electrolyte conditions. To further elucidate these structures, we calculate and visualize their formation free energies using density functional theory (DFT), combined with heat map analysis. Additionally, MLFF simulations extend our predictions to nanosecond-scale trajectories, surpassing the limitations of picosecond-scale AIMD. The predicted solvated structures show strong agreement with both AIMD and DFT results, demonstrating the robustness of our approach. Thus, by leveraging these comprehensive methods, we provide a more reliable framework for predicting solvation structures in calcium ion and other battery electrolytes.</p>\",\"PeriodicalId\":82,\"journal\":{\"name\":\"Journal of Materials Chemistry A\",\"volume\":\" 47\",\"pages\":\" 33150-33161\"},\"PeriodicalIF\":9.5000,\"publicationDate\":\"2024-10-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Materials Chemistry A\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/ta/d4ta06675h\",\"RegionNum\":2,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/ta/d4ta06675h","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
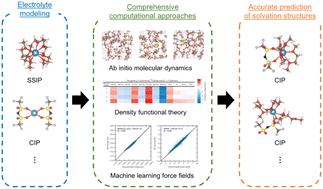
摘要
钙离子电池正在成为替代锂离子电池的一个关键重点。然而,在理解不同电解质物种如何影响其溶解结构方面仍存在关键差距。在本研究中,我们展示了一种综合预测方法,该方法整合了 ab initio 计算和机器学习力场 (MLFF),以应对这一挑战。利用 ab initio 分子动力学(AIMD)模拟,我们准确预测了第一溶壳内的溶解结构,同时还通过前沿轨道分析评估了它们的还原和氧化稳定性。这项分析比较了隐式和显式电解质条件。为了进一步阐明这些结构,我们使用密度泛函理论(DFT)并结合热图分析,计算并展示了它们的形成自由能。此外,MLFF 模拟将我们的预测扩展到纳秒级轨迹,超越了皮秒级 AIMD 的限制。预测的溶解结构与 AIMD 和 DFT 结果显示出很强的一致性,证明了我们方法的稳健性。因此,通过利用这些综合方法,我们为预测钙离子和其他电池电解质中的溶解结构提供了一个更可靠的框架。
Computational prediction of solvation structures in calcium battery electrolytes†
Calcium ion batteries are emerging as a key focus in the pursuit of alternatives to lithium-ion batteries. However, a crucial gap remains in understanding how different electrolyte species influence their solvation structures. In this study, we demonstrate a comprehensive predictive approach that integrates ab initio calculations and machine learning force fields (MLFFs) to address this challenge. Using ab initio molecular dynamics (AIMD) simulations, we accurately predict the solvation structures within the first solvation shell, while also evaluating their reductive and oxidative stability through frontier orbital analysis. This analysis compares both implicit and explicit electrolyte conditions. To further elucidate these structures, we calculate and visualize their formation free energies using density functional theory (DFT), combined with heat map analysis. Additionally, MLFF simulations extend our predictions to nanosecond-scale trajectories, surpassing the limitations of picosecond-scale AIMD. The predicted solvated structures show strong agreement with both AIMD and DFT results, demonstrating the robustness of our approach. Thus, by leveraging these comprehensive methods, we provide a more reliable framework for predicting solvation structures in calcium ion and other battery electrolytes.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


