萘吲哚-2-羧酰胺作为一种亲脂型 Hetarene-Anthraquinones 化合物,对具有 P-gp 抗性的肿瘤细胞有效
IF 6
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
对化疗产生多药耐药性(MDR)是成功治疗癌症的一大障碍。为了提高蒽醌类抗肿瘤化合物对 MDR 癌细胞的药效,我们采用合理的设计方法开发了新的杂芳烃融合蒽醌类化合物。将萘并[2,3-f]吲哚支架中的羧酰胺基团从 3 位移到 2 位,增加了衍生物的亲脂性和 P-glycoprotein (P-gp) 结合力,从而增强了它们规避 P-gp 介导的 MDR 的能力。为了验证计算结果,我们根据 2-炔基-3-氨基-1,4-二甲氧基蒽醌在温和碱性条件下使用四正丁基氟化铵 (TBAF) 进行 5-endo-dig 环化反应的结果,开发了一种杂环化为萘并[2,3-f]吲哚-2-羧酸酯的方案。合成的萘吲哚-2-羧酰胺类化合物,特别是羧酰胺片段中含有 (S)-3- 氨基吡咯烷的化合物 1a,显示出最高的抗增殖活性。最重要的是,1a 能抑制 P-gp 阳性的 K562/4 白血病肿瘤细胞系的生长(抗性指数 = 2.4),而其 3-异构体 LCTA-2640 和 Dox 则不能(抗性指数分别为 125 和 140)。对细胞内摄取和分布的研究表明,与 3-取代异构体不同,1a 能有效地在耐药性肿瘤细胞内积累,这证实了硅学数据与实验数据之间的相关性。先导化合物 1a 与 DNA 双链相互作用,抑制拓扑异构酶 1,但不会诱发氧化应激。用 1a 处理会增加 K562 和 K562/4 亚系的凋亡细胞数量,与细胞周期阶段无关。综上所述,这项工作提供了一个有趣的例子,说明化学结构上的微小变化如何导致抗肿瘤特性的显著差异。总之,我们发现了一类强效化合物,它们在抗击耐药性肿瘤细胞方面具有独特的优势。本文章由计算机程序翻译,如有差异,请以英文原文为准。
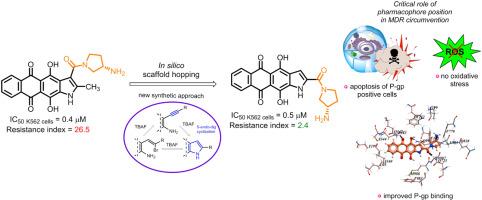
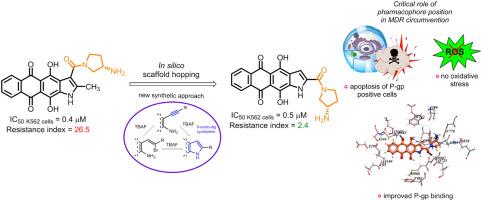
Naphthoindole-2-carboxamides as a lipophilic chemotype of hetarene-anthraquinones potent against P-gp resistant tumor cells
The acquisition of multidrug resistance (MDR) to chemotherapy is a major obstacle to successful cancer treatment. Aiming to improve the potency of anthraquinone-derived antitumor compounds against MDR cancer cells, we employed a rational design approach to develop new heteroarene-fused anthraquinones. Shifting the carboxamide group in the naphtho[2,3-f]indole scaffold from the 3-position to 2 increased the lipophilicity and P-glycoprotein (P-gp) binding of the derivatives, potentially enhancing their ability to circumvent P-gp-mediated MDR. To validate the computations, we developed a scheme for heterocyclization into esters of naphtho[2,3-f]indole-2-carboxylic acid, based on the 5-endo-dig cyclization of 2-alkynyl-3-amino-1,4-dimethoxyanthraquinone under mild basic conditions using tetra-n-butylammonium fluoride (TBAF). The synthesized naphthoindole-2-carboxamides, particularly compound 1a bearing (S)-3-aminopyrrolidine in the carboxamide fragment, demonstrated the highest antiproliferative activity. Most importantly, 1a suppressed the growth of the P-gp-positive K562/4 leukemia tumor cell line (resistance index = 2.4), while its 3-isomer LCTA-2640 and Dox did not (RI = 125 and 140, respectively). Studies of intracellular uptake and distribution showed that 1a, unlike its 3-substituted isomer, effectively accumulated in resistant tumor cells, confirming the correlation between in silico and experimental data. The lead compound 1a interacts with DNA duplex and inhibits topoisomerase 1 but does not induce oxidative stress. Treatment with 1a increases the population of apoptotic cells in both K562 and K562/4 sublines, regardless of the cell cycle phase. Taken together, this work provides an interesting example of how a little modification in chemical structure can lead to striking differences in antitumor properties. In conclusion, we have identified a potent class of compounds that offer distinct advantages in combating resistant tumor cells.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


