利用密度泛函理论进行无铅卤化物包晶材料的机器学习强化设计
IF 2.4
4区 物理与天体物理
Q3 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
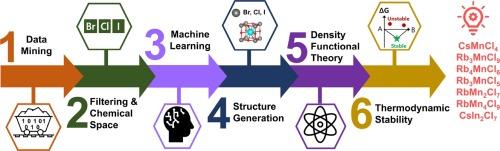
为了推动制造环境可持续的无铅过氧化物太阳能电池,对新兴的无毒过氧化物材料进行了研究。本研究介绍了一种机器学习方法,旨在预测有望用于光伏应用的创新型卤化物包晶材料。新预测的七种材料如下:CsMnCl4、Rb3Mn2Cl9、Rb4MnCl6、Rb3MnCl5、RbMn2Cl7、RbMn4Cl9 和 CsIn2Cl7。首先使用机器学习方法对预测的化合物进行筛选,然后通过密度泛函理论计算验证其有效性。CsMnCl4 是其中的佼佼者,它显示出 1.37 eV 的带隙,处于肖克利-奎塞尔极限之内,因此适合光伏应用。通过机器学习与密度泛函理论的结合,本研究提出了一种更有效、更全面的方法来发现和设计材料。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Machine learning-enhanced design of lead-free halide perovskite materials using density functional theory
The investigation of emerging non-toxic perovskite materials has been undertaken to advance the fabrication of environmentally sustainable lead-free perovskite solar cells. This study introduces a machine learning methodology aimed at predicting innovative halide perovskite materials that hold promise for use in photovoltaic applications. The seven newly predicted materials are as follows: CsMnCl4, Rb3Mn2Cl9, Rb4MnCl6, Rb3MnCl5, RbMn2Cl7, RbMn4Cl9, and CsIn2Cl7. The predicted compounds are first screened using a machine learning approach, and their validity is subsequently verified through density functional theory calculations. CsMnCl4 is notable among them, displaying a bandgap of 1.37 eV, falling within the Shockley-Queisser limit, making it suitable for photovoltaic applications. Through the integration of machine learning and density functional theory, this study presents a methodology that is more effective and thorough for the discovery and design of materials.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Current Applied Physics
物理-材料科学:综合
CiteScore
4.80
自引率
0.00%
发文量
213
审稿时长
33 days
期刊介绍:
Current Applied Physics (Curr. Appl. Phys.) is a monthly published international journal covering all the fields of applied science investigating the physics of the advanced materials for future applications.
Other areas covered: Experimental and theoretical aspects of advanced materials and devices dealing with synthesis or structural chemistry, physical and electronic properties, photonics, engineering applications, and uniquely pertinent measurement or analytical techniques.
Current Applied Physics, published since 2001, covers physics, chemistry and materials science, including bio-materials, with their engineering aspects. It is a truly interdisciplinary journal opening a forum for scientists of all related fields, a unique point of the journal discriminating it from other worldwide and/or Pacific Rim applied physics journals.
Regular research papers, letters and review articles with contents meeting the scope of the journal will be considered for publication after peer review.
The Journal is owned by the Korean Physical Society.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


