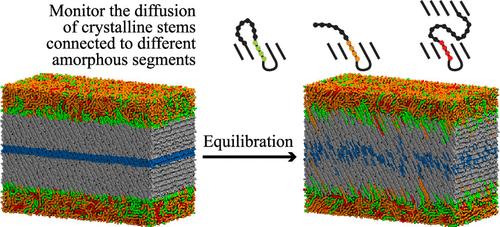
{"title":"结晶茎的热激活松弛对聚乙烯结晶/非晶界面分子拓扑结构的依赖性。","authors":"Yiyang Li, Jianlan Ye, Vipin Agrawal, Jay Oswald","doi":"10.1021/acs.jctc.4c00400","DOIUrl":null,"url":null,"abstract":"<p><p>We investigate the relaxation dynamics of crystalline stems in relation to the molecular topology of the crystalline/amorphous interface, employing coarse-grained molecular dynamics. To efficiently generate model semicrystalline systems of linear polyethylene with a realistic interphase morphology, we simplified the Monte Carlo method by introducing molecular dynamics for faster relaxation. The structural properties of the generated systems are validated against experimental measurements, theoretical predictions, and existing simulation data. The models suggest that the probability distribution of loop-entry sites on the lamellar surface can be described by a power law in terms of the distance between the entry sites. By considering realistic interphase morphology, we are able to improve the prediction of the overall activation energy for the relaxation of crystalline stems, aligning it closely with experimental measurements. The largest model predicts that crystalline stems connected via large loops, i.e., those that exceed the entanglement length, and long tails are associated with increased activation energy; whereas stems connected to shorter tails show the lowest activation energy. These predictions can guide the future development of tougher semicrystalline polymers by providing insights into how amorphous chain morphology contributes to the activation energy and the relaxation dynamics of crystalline chains.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-11-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Dependence of Thermally Activated Relaxation of Crystalline Stems on the Molecular Topology at Crystalline/Amorphous Interfaces in Polyethylene.\",\"authors\":\"Yiyang Li, Jianlan Ye, Vipin Agrawal, Jay Oswald\",\"doi\":\"10.1021/acs.jctc.4c00400\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We investigate the relaxation dynamics of crystalline stems in relation to the molecular topology of the crystalline/amorphous interface, employing coarse-grained molecular dynamics. To efficiently generate model semicrystalline systems of linear polyethylene with a realistic interphase morphology, we simplified the Monte Carlo method by introducing molecular dynamics for faster relaxation. The structural properties of the generated systems are validated against experimental measurements, theoretical predictions, and existing simulation data. The models suggest that the probability distribution of loop-entry sites on the lamellar surface can be described by a power law in terms of the distance between the entry sites. By considering realistic interphase morphology, we are able to improve the prediction of the overall activation energy for the relaxation of crystalline stems, aligning it closely with experimental measurements. The largest model predicts that crystalline stems connected via large loops, i.e., those that exceed the entanglement length, and long tails are associated with increased activation energy; whereas stems connected to shorter tails show the lowest activation energy. These predictions can guide the future development of tougher semicrystalline polymers by providing insights into how amorphous chain morphology contributes to the activation energy and the relaxation dynamics of crystalline chains.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-11-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00400\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00400","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/28 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Dependence of Thermally Activated Relaxation of Crystalline Stems on the Molecular Topology at Crystalline/Amorphous Interfaces in Polyethylene.
We investigate the relaxation dynamics of crystalline stems in relation to the molecular topology of the crystalline/amorphous interface, employing coarse-grained molecular dynamics. To efficiently generate model semicrystalline systems of linear polyethylene with a realistic interphase morphology, we simplified the Monte Carlo method by introducing molecular dynamics for faster relaxation. The structural properties of the generated systems are validated against experimental measurements, theoretical predictions, and existing simulation data. The models suggest that the probability distribution of loop-entry sites on the lamellar surface can be described by a power law in terms of the distance between the entry sites. By considering realistic interphase morphology, we are able to improve the prediction of the overall activation energy for the relaxation of crystalline stems, aligning it closely with experimental measurements. The largest model predicts that crystalline stems connected via large loops, i.e., those that exceed the entanglement length, and long tails are associated with increased activation energy; whereas stems connected to shorter tails show the lowest activation energy. These predictions can guide the future development of tougher semicrystalline polymers by providing insights into how amorphous chain morphology contributes to the activation energy and the relaxation dynamics of crystalline chains.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


