利用数据驱动的潜空间融合策略生成对抗网络进行晶体结构预测
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
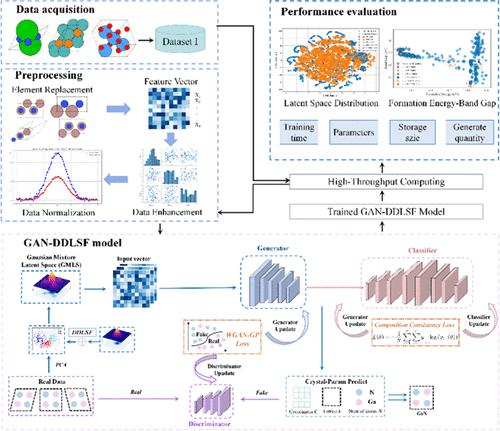
晶体结构预测(CSP)是材料设计的一个重要领域。在此,我们提出了一种新颖的生成对抗网络模型,该模型以数据驱动方法为指导,并结合了晶体的真实物理结构,以解决高维数据的复杂性并提高材料科学领域的预测精度。该模型被称为 GAN-DDLSF,它引入了一种名为数据驱动潜空间融合(DDLSF)的新型采样方法,旨在通过将真实数据的统计特性与标准高斯分布相结合来优化生成式对抗网络(GANs)的潜空间,从而有效缓解 GANs 中普遍存在的 "模式崩溃 "问题。我们的方法专门针对氮化镓(GaN)等二元晶体结构引入了更精细的生成机制。通过优化氮化镓的特定晶体学特征,同时保持结构的合理性,我们在预测和设计这一特定材料系统的结构时实现了更高的精度和效率。该模型生成了 9321 个氮化镓二元晶体结构,其中 16.59% 达到稳定状态,24.21% 属于可稳定状态。这些结果可以大大提高晶体结构预测的准确性,并为 GAN-DDLSF 方法发现和设计二元、三元和多元材料的潜力提供了宝贵的见解,为材料科学研究和应用提供了新的视角和方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Crystal Structure Prediction Using Generative Adversarial Network with Data-Driven Latent Space Fusion Strategy
Crystal structure prediction (CSP) is an important field of material design. Herein, we propose a novel generative adversarial network model, guided by a data-driven approach and incorporating the real physical structure of crystals, to address the complexity of high-dimensional data and improve prediction accuracy in materials science. The model, termed GAN-DDLSF, introduces a novel sampling method called data-driven latent space fusion (DDLSF), which aims to optimize the latent space of generative adversarial networks (GANs) by combining the statistical properties of real data with a standard Gaussian distribution, effectively mitigating the “mode collapse” problem prevalent in GANs. Our approach introduces a more refined generation mechanism specifically for binary crystal structures such as gallium nitride (GaN). By optimizing for the specific crystallographic features of GaN while maintaining structural rationality, we achieve higher precision and efficiency in predicting and designing structures for this particular material system. The model generates 9321 GaN binary crystal structures, with 16.59% reaching a stable state and 24.21% found to be metastable. These results can significantly enhance the accuracy of crystal structure predictions and provide valuable insights into the potential of the GAN-DDLSF approach for the discovery and design of binary, ternary, and multinary materials, offering new perspectives and methods for materials science research and applications.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


