利用机器学习和量子启发的相似性分析筛选高熵合金电催化剂
IF 17.3
1区 材料科学
Q1 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
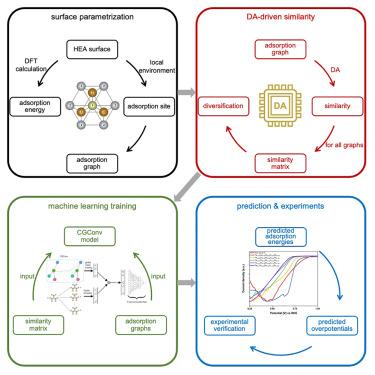
密度泛函理论(DFT)根据化学中间体在预期吸附位点上的能量计算过电势,有助于发现新的电催化剂。我们假设,在根据 DFT 数据训练机器学习模型时,可以通过引入吸附位点之间相似性的定量指标来提高准确性。当我们使用相似性作为输入特征来增强基于图神经网络的机器学习工作流程时,我们发现所需的训练数据集从 1600 个减少到 800 个,从而提高了 2 倍的速度:训练到一定准确度所需的 DFT 计算量减少了一半。这种方法确定了 Fe0.125Co0.125Ni0.229Ir0.229Ru0.292 是一种很有前途的氧还原反应催化剂,其过电势为 0.24 V,优于 Pt/C 基准。我们通过实验研究了另外四种 HEA,检验了计算方法的预测能力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
High-entropy alloy electrocatalysts screened using machine learning informed by quantum-inspired similarity analysis
The discovery of new electrocatalysts can be aided by density functional theory (DFT) computation of overpotentials based on the energies of chemical intermediates on prospective adsorption sites. We hypothesize that when training a machine learning model on DFT data, one could improve accuracy by introducing a quantitative measure of similarity among adsorption sites. When we augment a graph neural network-based machine learning workflow using similarity as an input feature, we find that the required training dataset size is decreased from 1,600 to 800, leading to a 2× acceleration: the number of DFT calculations required to train to a given level of accuracy is cut in half. This approach identifies Fe0.125Co0.125Ni0.229Ir0.229Ru0.292 as a promising oxygen reduction reaction catalyst with an overpotential of 0.24 V, outperforming a Pt/C benchmark. We examine, by studying experimentally four additional HEAs, the predictive power of the computational approach.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Matter
MATERIALS SCIENCE, MULTIDISCIPLINARY-
CiteScore
26.30
自引率
2.60%
发文量
367
期刊介绍:
Matter, a monthly journal affiliated with Cell, spans the broad field of materials science from nano to macro levels,covering fundamentals to applications. Embracing groundbreaking technologies,it includes full-length research articles,reviews, perspectives,previews, opinions, personnel stories, and general editorial content.
Matter aims to be the primary resource for researchers in academia and industry, inspiring the next generation of materials scientists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


