用于单细胞 RNA 测序数据差异表达分析的矩方法框架
IF 45.5
1区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
单细胞 RNA 测序(scRNA-seq)数据的差异表达分析是描述实验因素如何影响基因表达分布的核心。然而,区分细胞间变异性的生物和技术来源以及评估细胞组间定量比较的统计意义仍具有挑战性。我们介绍了 Memento,这是一种对 scRNA-seq 数据的平均表达、变异性和基因相关性进行稳健而高效的差异分析的工具,可扩展至数百万个细胞和数千个样本。我们将Memento应用于7万个气管上皮细胞,以鉴定干扰素反应基因;16万个CRISPR-Cas9扰动T细胞,以重建基因调控网络;120万个外周血单核细胞(PBMC),以绘制细胞类型特异性定量性状位点(QTL);以及5000万个细胞的CELLxGENE Discover语料库,以比较任意细胞群。在所有情况下,与现有方法相比,Memento 在平均表达方面都能发现更显著、更可重复的差异。它还发现了变异性和基因相关性的差异,这表明扰动带来了不同的转录调控机制。本文章由计算机程序翻译,如有差异,请以英文原文为准。
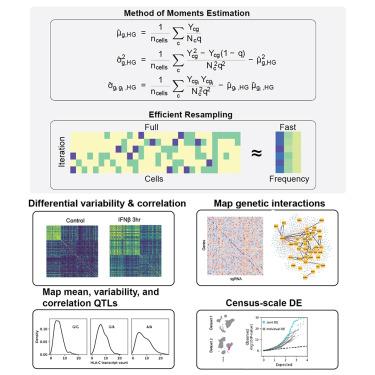
Method of moments framework for differential expression analysis of single-cell RNA sequencing data
Differential expression analysis of single-cell RNA sequencing (scRNA-seq) data is central for characterizing how experimental factors affect the distribution of gene expression. However, distinguishing between biological and technical sources of cell-cell variability and assessing the statistical significance of quantitative comparisons between cell groups remain challenging. We introduce Memento, a tool for robust and efficient differential analysis of mean expression, variability, and gene correlation from scRNA-seq data, scalable to millions of cells and thousands of samples. We applied Memento to 70,000 tracheal epithelial cells to identify interferon-responsive genes, 160,000 CRISPR-Cas9 perturbed T cells to reconstruct gene-regulatory networks, 1.2 million peripheral blood mononuclear cells (PBMCs) to map cell-type-specific quantitative trait loci (QTLs), and the 50-million-cell CELLxGENE Discover corpus to compare arbitrary cell groups. In all cases, Memento identified more significant and reproducible differences in mean expression compared with existing methods. It also identified differences in variability and gene correlation that suggest distinct transcriptional regulation mechanisms imparted by perturbations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Cell
生物-生化与分子生物学
CiteScore
110.00
自引率
0.80%
发文量
396
审稿时长
2 months
期刊介绍:
Cells is an international, peer-reviewed, open access journal that focuses on cell biology, molecular biology, and biophysics. It is affiliated with several societies, including the Spanish Society for Biochemistry and Molecular Biology (SEBBM), Nordic Autophagy Society (NAS), Spanish Society of Hematology and Hemotherapy (SEHH), and Society for Regenerative Medicine (Russian Federation) (RPO).
The journal publishes research findings of significant importance in various areas of experimental biology, such as cell biology, molecular biology, neuroscience, immunology, virology, microbiology, cancer, human genetics, systems biology, signaling, and disease mechanisms and therapeutics. The primary criterion for considering papers is whether the results contribute to significant conceptual advances or raise thought-provoking questions and hypotheses related to interesting and important biological inquiries.
In addition to primary research articles presented in four formats, Cells also features review and opinion articles in its "leading edge" section, discussing recent research advancements and topics of interest to its wide readership.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


