Jeyanthi Suppiah, Saiful Safuan Md Sani, Safiah Sabrina Hassan, Nur Iman Fasohah Nadzar, Nurul 'Izzah Ibrahim, Ravindran Thayan, Rozainanee Mohd Zain
{"title":"通过 RNA 测序揭示登革热感染的潜在基因生物标志物。","authors":"Jeyanthi Suppiah, Saiful Safuan Md Sani, Safiah Sabrina Hassan, Nur Iman Fasohah Nadzar, Nurul 'Izzah Ibrahim, Ravindran Thayan, Rozainanee Mohd Zain","doi":"10.1007/s11262-024-02114-2","DOIUrl":null,"url":null,"abstract":"<p><p>Dengue virus hijacks host cell mechanisms and immune responses in order to replicate efficiently. The interaction between the host and the virus affects the host's gene expression, which remains largely unexplored. This pilot study aimed to profile the host transcriptome as a potential strategy for identifying specific biomarkers for dengue prediction and detection. High-throughput RNA sequencing (RNA-seq) was employed to generate host transcriptome profiles in 16 dengue patients and 10 healthy controls. Differentially expressed genes (DEGs) were identified in patients with severe dengue and those with dengue with warning signs compared to healthy individuals. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to elucidate the functions of upregulated and downregulated genes. Compared to healthy controls, 6466 genes were significantly differentially expressed (p < 0.05) in the dengue with warning signs group and 3082 genes in the severe dengue group, with over half being upregulated. The major KEGG pathways implicated included transport and catabolism (14.4%-16.3%), signal transduction (6.6%-7.3%), global and overview maps (6.7%-7.1%), viral diseases (4.6%-4.8%), and the immune system (4.4%-4.6%). Several genes exhibited consistent and significant upregulation across all dengue patients, regardless of severity: Interferon alpha inducible protein 27 (IFI27), Potassium Channel Tetramerization Domain Containing 14 (KCTD14), Syndecan 1 (SDC1), DCC netrin 1 receptor (DCC), Ubiquitin C-terminal hydrolase L1 (UCHL1), Marginal zone B and B1 cell-specific protein (MZB1), Nestin (NES), C-C motif chemokine ligand 2 (CCL2), TNF receptor superfamily member 17 (TNFSF17), and TNF receptor superfamily member 13B (TNFRSF13B). Further analysis revealed potential biomarkers for severe dengue prediction, including TNF superfamily member 15 (TNFSF15), Plasminogen Activator Inhibitor-2 (SERPINB2), motif chemokine ligand 7 (CCL7), aconitate decarboxylase 1 (ACOD1), Metallothionein 1G (MT1G), and Myosin Light Chain Kinase (MYLK2), which were expressed 3.5 times, 2.9 times, 2.3 times, 2.1 times, 1.7 times, and 1.4 times greater, respectively, than dengue patients exhibiting warning signs. The identification of these host biomarkers through RNA-sequencing holds promising implications and potential to augment existing dengue detection algorithms, contributing significantly to improved diagnostic and prognostic capabilities.</p>","PeriodicalId":51212,"journal":{"name":"Virus Genes","volume":" ","pages":"26-37"},"PeriodicalIF":1.9000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11787201/pdf/","citationCount":"0","resultStr":"{\"title\":\"Unraveling potential gene biomarkers for dengue infection through RNA sequencing.\",\"authors\":\"Jeyanthi Suppiah, Saiful Safuan Md Sani, Safiah Sabrina Hassan, Nur Iman Fasohah Nadzar, Nurul 'Izzah Ibrahim, Ravindran Thayan, Rozainanee Mohd Zain\",\"doi\":\"10.1007/s11262-024-02114-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Dengue virus hijacks host cell mechanisms and immune responses in order to replicate efficiently. The interaction between the host and the virus affects the host's gene expression, which remains largely unexplored. This pilot study aimed to profile the host transcriptome as a potential strategy for identifying specific biomarkers for dengue prediction and detection. High-throughput RNA sequencing (RNA-seq) was employed to generate host transcriptome profiles in 16 dengue patients and 10 healthy controls. Differentially expressed genes (DEGs) were identified in patients with severe dengue and those with dengue with warning signs compared to healthy individuals. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to elucidate the functions of upregulated and downregulated genes. Compared to healthy controls, 6466 genes were significantly differentially expressed (p < 0.05) in the dengue with warning signs group and 3082 genes in the severe dengue group, with over half being upregulated. The major KEGG pathways implicated included transport and catabolism (14.4%-16.3%), signal transduction (6.6%-7.3%), global and overview maps (6.7%-7.1%), viral diseases (4.6%-4.8%), and the immune system (4.4%-4.6%). Several genes exhibited consistent and significant upregulation across all dengue patients, regardless of severity: Interferon alpha inducible protein 27 (IFI27), Potassium Channel Tetramerization Domain Containing 14 (KCTD14), Syndecan 1 (SDC1), DCC netrin 1 receptor (DCC), Ubiquitin C-terminal hydrolase L1 (UCHL1), Marginal zone B and B1 cell-specific protein (MZB1), Nestin (NES), C-C motif chemokine ligand 2 (CCL2), TNF receptor superfamily member 17 (TNFSF17), and TNF receptor superfamily member 13B (TNFRSF13B). Further analysis revealed potential biomarkers for severe dengue prediction, including TNF superfamily member 15 (TNFSF15), Plasminogen Activator Inhibitor-2 (SERPINB2), motif chemokine ligand 7 (CCL7), aconitate decarboxylase 1 (ACOD1), Metallothionein 1G (MT1G), and Myosin Light Chain Kinase (MYLK2), which were expressed 3.5 times, 2.9 times, 2.3 times, 2.1 times, 1.7 times, and 1.4 times greater, respectively, than dengue patients exhibiting warning signs. The identification of these host biomarkers through RNA-sequencing holds promising implications and potential to augment existing dengue detection algorithms, contributing significantly to improved diagnostic and prognostic capabilities.</p>\",\"PeriodicalId\":51212,\"journal\":{\"name\":\"Virus Genes\",\"volume\":\" \",\"pages\":\"26-37\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2025-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11787201/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Virus Genes\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s11262-024-02114-2\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/14 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Virus Genes","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11262-024-02114-2","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/14 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Unraveling potential gene biomarkers for dengue infection through RNA sequencing.
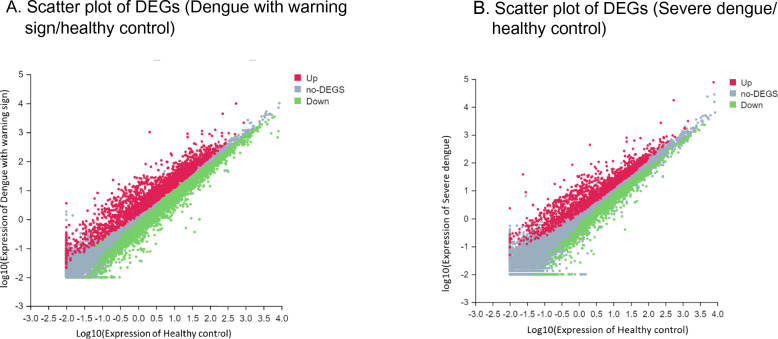
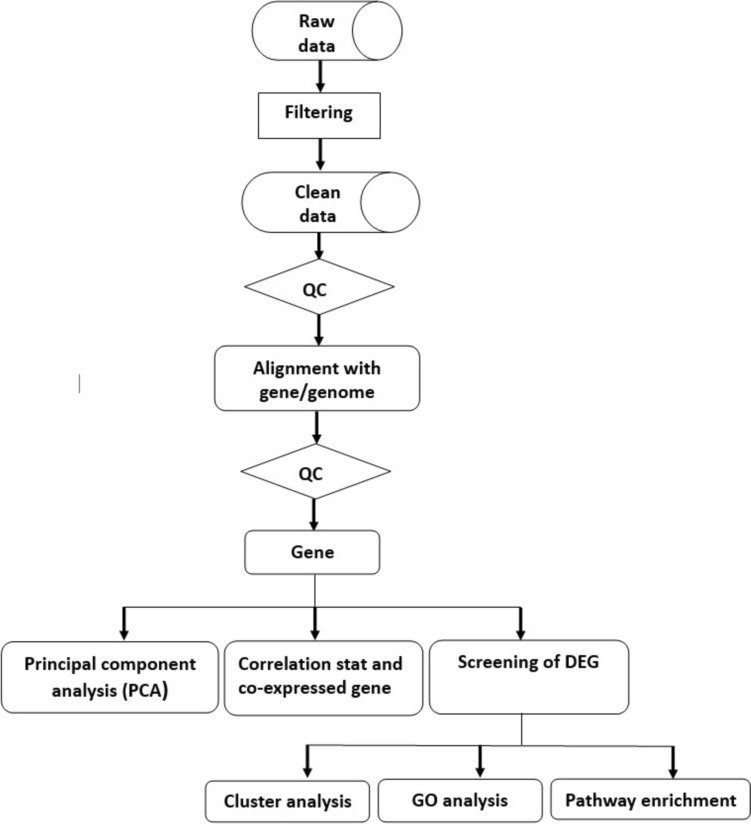
Dengue virus hijacks host cell mechanisms and immune responses in order to replicate efficiently. The interaction between the host and the virus affects the host's gene expression, which remains largely unexplored. This pilot study aimed to profile the host transcriptome as a potential strategy for identifying specific biomarkers for dengue prediction and detection. High-throughput RNA sequencing (RNA-seq) was employed to generate host transcriptome profiles in 16 dengue patients and 10 healthy controls. Differentially expressed genes (DEGs) were identified in patients with severe dengue and those with dengue with warning signs compared to healthy individuals. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to elucidate the functions of upregulated and downregulated genes. Compared to healthy controls, 6466 genes were significantly differentially expressed (p < 0.05) in the dengue with warning signs group and 3082 genes in the severe dengue group, with over half being upregulated. The major KEGG pathways implicated included transport and catabolism (14.4%-16.3%), signal transduction (6.6%-7.3%), global and overview maps (6.7%-7.1%), viral diseases (4.6%-4.8%), and the immune system (4.4%-4.6%). Several genes exhibited consistent and significant upregulation across all dengue patients, regardless of severity: Interferon alpha inducible protein 27 (IFI27), Potassium Channel Tetramerization Domain Containing 14 (KCTD14), Syndecan 1 (SDC1), DCC netrin 1 receptor (DCC), Ubiquitin C-terminal hydrolase L1 (UCHL1), Marginal zone B and B1 cell-specific protein (MZB1), Nestin (NES), C-C motif chemokine ligand 2 (CCL2), TNF receptor superfamily member 17 (TNFSF17), and TNF receptor superfamily member 13B (TNFRSF13B). Further analysis revealed potential biomarkers for severe dengue prediction, including TNF superfamily member 15 (TNFSF15), Plasminogen Activator Inhibitor-2 (SERPINB2), motif chemokine ligand 7 (CCL7), aconitate decarboxylase 1 (ACOD1), Metallothionein 1G (MT1G), and Myosin Light Chain Kinase (MYLK2), which were expressed 3.5 times, 2.9 times, 2.3 times, 2.1 times, 1.7 times, and 1.4 times greater, respectively, than dengue patients exhibiting warning signs. The identification of these host biomarkers through RNA-sequencing holds promising implications and potential to augment existing dengue detection algorithms, contributing significantly to improved diagnostic and prognostic capabilities.
期刊介绍:
Viruses are convenient models for the elucidation of life processes. The study of viruses is again on the cutting edge of biological sciences: systems biology, genomics, proteomics, metagenomics, using the newest most powerful tools.
Huge amounts of new details on virus interactions with the cell, other pathogens and the hosts – animal (including human), insect, fungal, plant, bacterial, and archaeal - and their role in infection and disease are forthcoming in perplexing details requiring analysis and comments.
Virus Genes is dedicated to the publication of studies on the structure and function of viruses and their genes, the molecular and systems interactions with the host and all applications derived thereof, providing a forum for the analysis of data and discussion of its implications, and the development of new hypotheses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


