Weikang Xian, Amitesh Maiti, Andrew P. Saab and Ying Li
{"title":"聚(二甲基-共-二苯基)硅氧烷粗粒度分子动力学模型的开发。","authors":"Weikang Xian, Amitesh Maiti, Andrew P. Saab and Ying Li","doi":"10.1039/D4SM00875H","DOIUrl":null,"url":null,"abstract":"<p >Polydimethylsiloxane is an important polymeric material with a wide range of applications. However, environmental effects like low temperature can induce crystallization in this material with resulting changes in its structural and dynamic properties. The incorporation of phenyl-siloxane components, <em>e.g.</em>, as in a poly(dimethyl-<em>co</em>-diphenyl)siloxane random copolymer, is known to suppress such crystallization. Molecular dynamics (MD) simulations can be a powerful tool to understand such effects in atomistic detail. Unfortunately, all-atomistic molecular dynamics (AAMD) is limited in both spatial dimensions and simulation times it can probe. To overcome such constraints and to extend to more useful length- and time-scales, we systematically develop a coarse-grained molecular dynamics (CGMD) model for the poly(dimethyl-<em>co</em>-diphenyl)siloxane system with bonded and non-bonded interactions determined from all-atomistic simulations by the iterative Boltzmann inversion (IBI) method. Additionally, we propose a lever rule that can be useful to generate non-bonded potentials for such systems without reference to the all-atomistic ground truth. Our model captures the structural and dynamic properties of the copolymer material with quantitative accuracy and is useful to study long-time dynamics of highly-entangled systems, sequence-dependent properties, phase behaviour, <em>etc.</em></p>","PeriodicalId":103,"journal":{"name":"Soft Matter","volume":" 42","pages":" 8480-8492"},"PeriodicalIF":2.9000,"publicationDate":"2024-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Development of a coarse-grained molecular dynamics model for poly(dimethyl-co-diphenyl)siloxane†\",\"authors\":\"Weikang Xian, Amitesh Maiti, Andrew P. Saab and Ying Li\",\"doi\":\"10.1039/D4SM00875H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Polydimethylsiloxane is an important polymeric material with a wide range of applications. However, environmental effects like low temperature can induce crystallization in this material with resulting changes in its structural and dynamic properties. The incorporation of phenyl-siloxane components, <em>e.g.</em>, as in a poly(dimethyl-<em>co</em>-diphenyl)siloxane random copolymer, is known to suppress such crystallization. Molecular dynamics (MD) simulations can be a powerful tool to understand such effects in atomistic detail. Unfortunately, all-atomistic molecular dynamics (AAMD) is limited in both spatial dimensions and simulation times it can probe. To overcome such constraints and to extend to more useful length- and time-scales, we systematically develop a coarse-grained molecular dynamics (CGMD) model for the poly(dimethyl-<em>co</em>-diphenyl)siloxane system with bonded and non-bonded interactions determined from all-atomistic simulations by the iterative Boltzmann inversion (IBI) method. Additionally, we propose a lever rule that can be useful to generate non-bonded potentials for such systems without reference to the all-atomistic ground truth. Our model captures the structural and dynamic properties of the copolymer material with quantitative accuracy and is useful to study long-time dynamics of highly-entangled systems, sequence-dependent properties, phase behaviour, <em>etc.</em></p>\",\"PeriodicalId\":103,\"journal\":{\"name\":\"Soft Matter\",\"volume\":\" 42\",\"pages\":\" 8480-8492\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-10-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Soft Matter\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/sm/d4sm00875h\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Soft Matter","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/sm/d4sm00875h","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
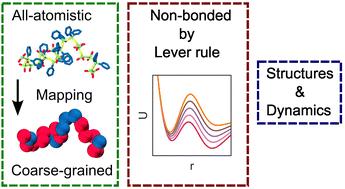
Development of a coarse-grained molecular dynamics model for poly(dimethyl-co-diphenyl)siloxane†
Polydimethylsiloxane is an important polymeric material with a wide range of applications. However, environmental effects like low temperature can induce crystallization in this material with resulting changes in its structural and dynamic properties. The incorporation of phenyl-siloxane components, e.g., as in a poly(dimethyl-co-diphenyl)siloxane random copolymer, is known to suppress such crystallization. Molecular dynamics (MD) simulations can be a powerful tool to understand such effects in atomistic detail. Unfortunately, all-atomistic molecular dynamics (AAMD) is limited in both spatial dimensions and simulation times it can probe. To overcome such constraints and to extend to more useful length- and time-scales, we systematically develop a coarse-grained molecular dynamics (CGMD) model for the poly(dimethyl-co-diphenyl)siloxane system with bonded and non-bonded interactions determined from all-atomistic simulations by the iterative Boltzmann inversion (IBI) method. Additionally, we propose a lever rule that can be useful to generate non-bonded potentials for such systems without reference to the all-atomistic ground truth. Our model captures the structural and dynamic properties of the copolymer material with quantitative accuracy and is useful to study long-time dynamics of highly-entangled systems, sequence-dependent properties, phase behaviour, etc.
期刊介绍:
Soft Matter is an international journal published by the Royal Society of Chemistry using Engineering-Materials Science: A Synthesis as its research focus. It publishes original research articles, review articles, and synthesis articles related to this field, reporting the latest discoveries in the relevant theoretical, practical, and applied disciplines in a timely manner, and aims to promote the rapid exchange of scientific information in this subject area. The journal is an open access journal. The journal is an open access journal and has not been placed on the alert list in the last three years.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


