水的可转移原子团簇扩展的高效参数化
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
我们利用原子团簇扩展(ACE)提出了一种高精度、可转移的水参数化方法。为了高效采样液态水,我们提出了一种新方法,即对各种冰相进行静态采样计算,并利用基于 ACE 的 D-optimality 算法的主动学习(AL)功能来选择相关的液态水构型,从而绕过计算密集的原子分子动力学模拟。我们的研究结果表明,ACE 描述因子使最初仅拟合冰结构的势能得以拟合,而后通过 AL 识别出的少量液态构型对其进行拟合,从而提供了对液态水的出色描述。所开发的电位与第一原理参考具有显著的一致性,能准确捕捉液态水的各种特性,包括结构特性(如配对相关函数、共价键剖面和氢键剖面)、动态特性(如振动态密度、扩散系数)和热力学特性(如冰的熔点 Ih)。我们的研究为水模拟中的机器学习势引入了一种新的高效采样技术,同时还提出了一种可转移的水原子间势,揭示了第一原理参考的准确性。这一进展不仅增强了我们对冰和液态水在原子水平上的关系的理解,还为研究复杂的水系统开辟了新的途径。本文章由计算机程序翻译,如有差异,请以英文原文为准。
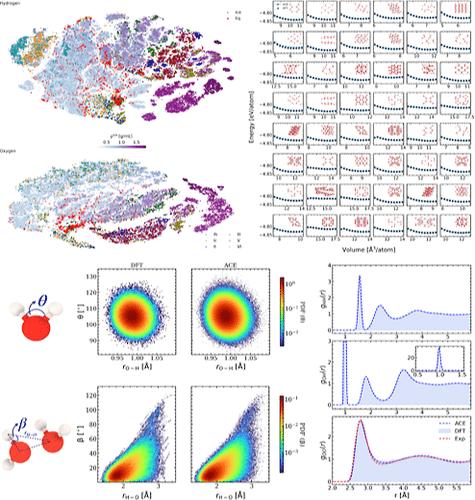
Efficient Parametrization of Transferable Atomic Cluster Expansion for Water
We present a highly accurate and transferable parametrization of water using the atomic cluster expansion (ACE). To efficiently sample liquid water, we propose a novel approach that involves sampling static calculations of various ice phases and utilizing the active learning (AL) feature of the ACE-based D-optimality algorithm to select relevant liquid water configurations, bypassing computationally intensive ab initio molecular dynamics simulations. Our results demonstrate that the ACE descriptors enable a potential initially fitted solely on ice structures, which is later upfitted with few configurations of liquid, identified with AL to provide an excellent description of liquid water. The developed potential exhibits remarkable agreement with first-principles reference, accurately capturing various properties of liquid water, including structural characteristics such as pair correlation functions, covalent bonding profiles, and hydrogen bonding profiles, as well as dynamic properties like the vibrational density of states, diffusion coefficient, and thermodynamic properties such as the melting point of the ice Ih. Our research introduces a new and efficient sampling technique for machine learning potentials in water simulations while also presenting a transferable interatomic potential for water that reveals the accuracy of first-principles reference. This advancement not only enhances our understanding of the relationship between ice and liquid water at the atomic level but also opens up new avenues for studying complex aqueous systems.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


