通过 D3S 为密度泛函理论提供更平滑的半经典色散:理解和解决 D3 色散校正模型中的非物理极小值
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
在密度泛函理论(DFT)中考虑伦敦色散相互作用的最广泛使用和计算效率最高的模型之一是 D3 色散修正模型。在这项研究中,我们证明了当原子配位数发生变化时,该模型会在势能面(PES)上诱发非物理最小值的出现。根据这些伪结构进行优化会导致在确定两个分子间的相互作用能和估算系统的热力学性质时出现重大误差。在几个具体的例子中,如 Kuratowski 型 H2-NiKur 和 H2-PdKur 团簇,这些局部最小值表现出极高的 PES 曲率,导致对谐波频率的错误估计以及对零点能和焓值的显著高估。虽然 D3 模型的这种错误行为相对罕见,但它可能发生在广泛的化学物种中,包括[Li(C6H6)]+ 复合物和二螺(吖啶)取代芘(DSAP)分子等分子。我们的分析表明,问题的根源在于 D3 模型中与 AB 原子对相关的 C6AB 系数的定义。为了解决这个问题,我们提出了对 D3 模型重新参数化的建议,即引入一种修改后的 C6AB 函数形式,该形式现在取决于所考虑的特定原子对。这种新模型被称为 D3-Smooth(简称 D3S),旨在平滑与色散修正相关的 PES。通过这种方法,我们证明 D3S 可以消除非物理的局部极小值,同时在相互作用能量基准集中保持母 D3 方法相当令人满意的精度。例如,在包含近 5000 个数据点的大型 MGCDB84 数据集中,使用 D3(BJ) 修正 B3LYP 与使用 D3S(BJ) 修正 B3LYP 的均方根差异仅为 0.12 kJ/mol。测试的其他 D3 校正函数也得到了类似的结果。与这一结果相一致的是,通过重新优化阻尼函数,B3LYP-D3S(0) 校正结果并没有得到明显改善。本文章由计算机程序翻译,如有差异,请以英文原文为准。
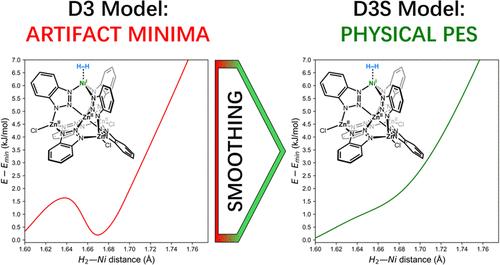
Smoother Semiclassical Dispersion for Density Functional Theory via D3S: Understanding and Addressing Unphysical Minima in the D3 Dispersion Correction Model
One of the most widely used and computationally efficient models that accounts for London dispersion interactions within density functional theory (DFT) is the D3 dispersion correction model. In this work, we demonstrate that this model can induce the appearance of unphysical minima on the potential energy surface (PES) when the coordination number of atoms changes. Optimizing to these artifactual structures can lead to significant errors in determining the interaction energy between two molecules and in estimating the thermodynamic properties of the system. In several specific examples, such as Kuratowski-type H2–NiKur and H2–PdKur clusters, these local minima exhibited extremely high PES curvature, resulting in incorrect estimations of harmonic frequencies and significant overestimations of zero-point energy and enthalpy values. Although such erroneous behavior of the D3 model is relatively rare, it can occur across a wide range of chemical species, including molecules like the [Li(C6H6)]+ complex and the dispiro(acridan)-substituted pyracene (DSAP) molecule. Our analysis reveals that the root of the problem lies in the definition of the AB atomic-pair dependent C6AB coefficients in the D3 model. To address this issue, we propose a reparameterization of the D3 model by introducing a modified C6AB functional form that now depends on the specific pair of considered atoms. This new model, termed D3-Smooth (or D3S for short), is designed to smooth out the PES associated with the dispersion correction. By doing so, we demonstrate that D3S eliminates unphysical local minima while maintaining the quite satisfactory accuracy of the parent D3 method in interaction energy benchmark sets. For example, the RMS difference between using the D3(BJ) correction to B3LYP and the D3S(BJ) correction across the large MGCDB84 data set of nearly 5000 data points is only 0.12 kJ/mol. Similar results are obtained for every other D3-corrected functional tested. Consistent with this result, no significant improvement could be obtained for the B3LYP-D3S(0) correction by reoptimizing the damping function.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


